
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- in China
- within Food, Drugs, Healthcare, Life Sciences, Technology and Insurance topic(s)
自2013年以来,国家药品监督管理局药品审评中心(下称 CDE)每年均会发布上一年的年度药品审评报告,对其上一年中药品审评工作的开展情况进行总结,报告逐渐覆盖药品注册申请受理情况、药品注册申请审评审批情况、加速通道建设和沟通交流开展情况、药品注册申请中存在的问题分析、重点治疗领域品种信息和中药审评审批情况等重点内容。
我们研读了2015年至2021年的年度药品审评报告,希望能够通过综合分析历年的年度药品审评报告,梳理出近年来CDE的主要工作情况和全国药品注册相关的形势,以此展望药品审评审批制度的改革情况与发展趋势,为未来的药品注册工作指引方向。
一、历年制度改革重点
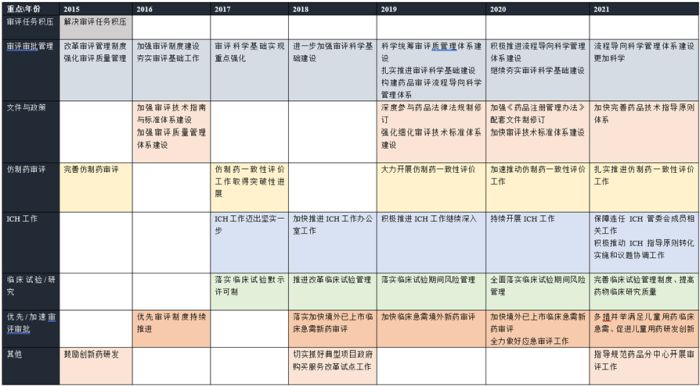
在历年的年度药品审评报告中,CDE均会对其在该年度中的主要工作措施及进展情况进行列举说明,从中可以看出CDE的工作重点变化,以及推进药品审评审批制度改革举措的集中区域。
下图提炼了近7年来年度药品审评报告中所提及的部分改革关键词。其中,解决审评任务积压是2015年的工作重点;而仿制药审评(出现五次)、ICH工作(出现五次)、临床试验管理(出现五次)、优先/加速审评审批(出现五次)等,是近六年反复多次出现的关键词。在本文的中篇及下篇中,我们将进一步结合以上的历年制度改革重点,从审评审批时限管理、化学药品注册分类变化与申请变化、仿制药一致性评价、生物制品注册发展、儿童用药与罕见病药物审评审批工作五大主题出发,解读近七年药品审评审批制度的发展。
二、药品注册申请存在的主要问题
在《2020年度药品审评报告》和《2021年度药品审评报告》中,CDE对一年中药品注册申请存在的主要问题进行了列举和分析。本文将对此进行梳理,从而希望对参与药物研发、注册的相关主体提供帮助和建议。
(一) 主要问题及分析
《2020年度药品审评报告》和《2021年度药品审评报告》分别从不同申请的分类角度和研发立题方面、有效性方面、安全性方面、质量可控性方面、合规性等方面归纳总结了药品注册申请存在的主要问题。
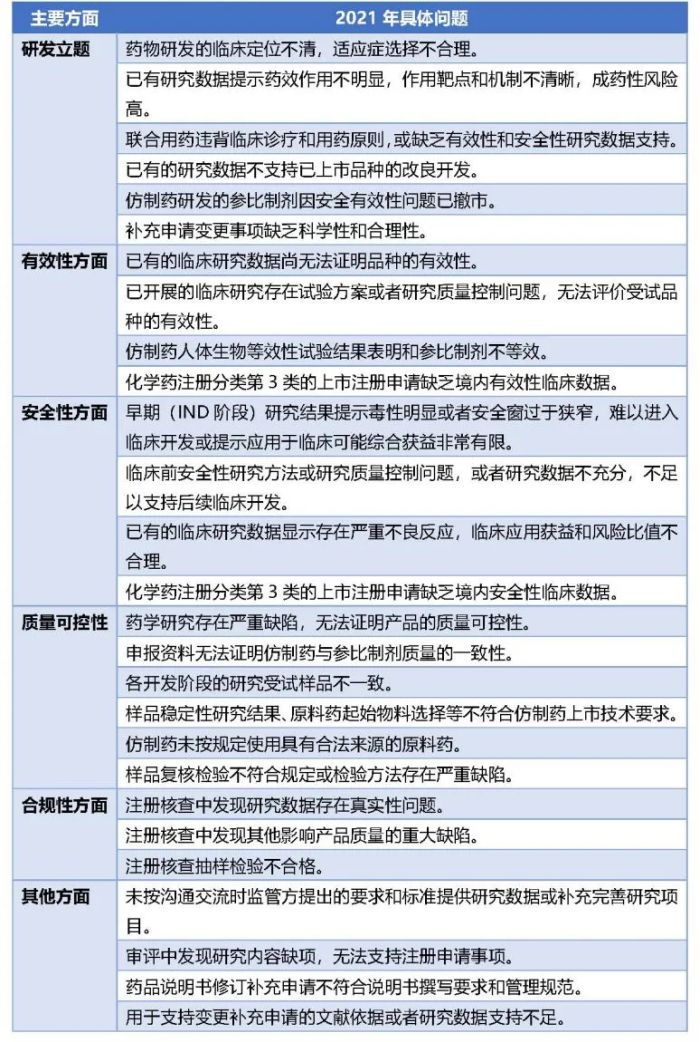
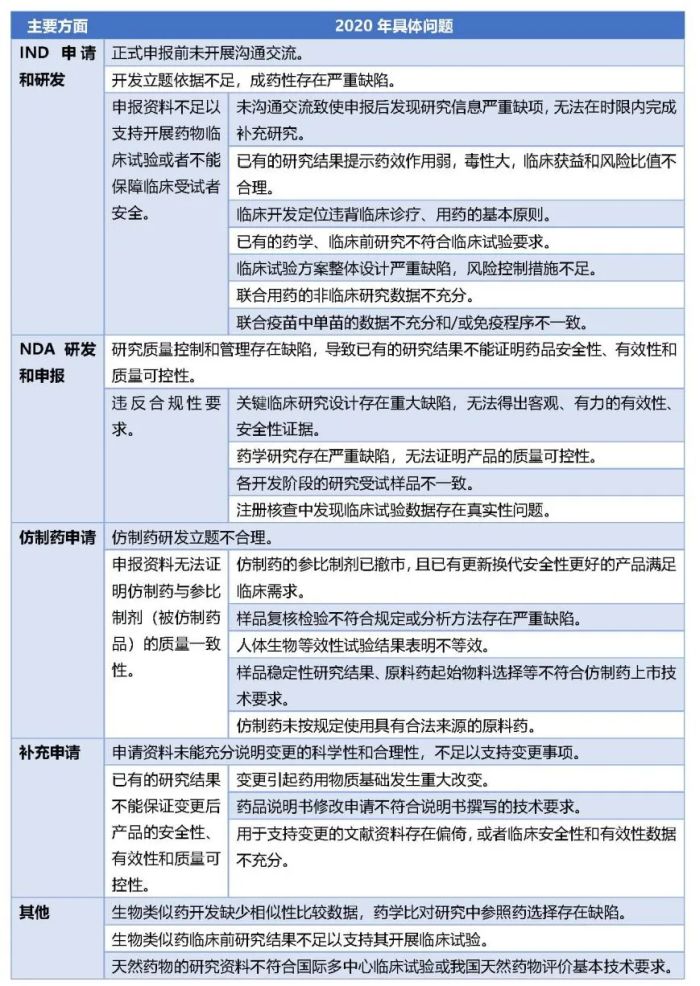
根据《2021年度药品审评报告》中的数据,2021年,药品注册申请经技术审评后审评结论为不批准/建议不批准的注册申请542件,其中,66.3%属于因申请人未能在规定时限内补充资料的情形;33.7%主要存在申报资料无法证明申请注册药品的安全性、有效性或质量问题。CDE指出,总体来看,2021年的注册申请中存在的主要问题,在分类、具体表现等方面与往年具有较大的相似性,但是也出现了一些变化,值得我们的关注:
首先,2021年出现的新问题包括申请人未按在临床试验申请前沟通交流时监管方提出的补充资料要求提交研究资料,导致审评过程中发现IND研究内容缺项。根据现行《药品注册管理办法》第88条的规定,申请人在药物临床试验申请的审评期间不得补充新的技术资料。因此,若申请人在提交注册申请资料时忽视了沟通交流中已明确的应提交的研究资料,将导致审评不通过。
其次,2020年列出的主要问题12个、具体问题19个,2021年列出的主要问题共28个,未列具体问题,虽然问题分类口径不一致,但是药监局表示在2021年基于某些问题而不批准的药品品种数量发生变化。一是2021年没有出现因未进行沟通交流而不批准的注册申请;二是因缺乏境内有效性、安全性临床数据而未获批准的化学药注册分类第3类上市注册申请数量较往年明显增加;三是因合规性问题而未获批准的注册申请数量较往年有减少趋势;四是因开发立题合理性问题未获批准的注册申请数量增加趋势明显。
(二) 启示和建议
结合药品审评报告中CDE对于前述问题的梳理分析,参与药物研发、注册的相关方可参考如下几点建议:
首先,应当充分重视药物开发的立题依据。近年来,相关方对于药品注册申请相关合规问题的重视程度不断提升,不合规现象相对减少。然而,除合规红线外,随着CDE对于药物研发质量要求的提升,药物研发的立题合理性问题在新形势下必须得到各方的重视。报告明确指出,药物开发应立足于临床需求,尤其应重视解决未被满足的临床需求问题;应以临床价值为导向,充分重视同类创新药开发的优势问题,避免群体化、低水平、重复性创新;应充分评估改良型新药的临床价值和优势;变更补充申请应遵循必要性与合理性原则等。
其次,应当妥善利用沟通交流机制。相关方应意识到,沟通交流机制不仅为药品注册申请提供了重要的便利,也可能对药品注册申请增设具体的监管要求。一方面,如报告指出,申请人可以充分利用现有的沟通交流机制,加强在药物开发关键环节与其他各环节与监管部门的沟通交流,以便消除信息不对等,达成问题共识,进而便利注册申请工作;另一方面,沟通交流过程中监管部门提出补充资料等发补监管要求必须得到相关主体的重视和遵循,以免影响注册申请结果。
再次,应当随时关注监管机关对于药品注册申报资料要求的更新变化。如前文所述,现行《药品注册管理办法》实施后,第3类化学药上市注册申请的管理要求发生调整,2020年《境外已上市境内未上市药品临床技术要求》明确规定了第3类化学药品上市注册的临床试验要求。及时把握监管部门对于药品注册分类和药品注册申报资料等方面的最新监管要求,有助于避免因缺乏境内有效性、安全性临床数据而导致第3类化学药未通过审批等问题的发生。
最后,应当加强创新药物开发的前期基础研究。报告提到,某些新机制、新靶点宜做充分的成药性评估,开展尽可能多的概念验证研究,以降低后续开发风险,以免造成研究资源浪费;创新药商业开发策略应建立在科学性基础上,重视成药性证据链的完整性;应遵循药物开发的科学逻辑,循序渐进,尽量减少非科学因素对开发进程的干扰。
以上是我们对2015年至2021年间年度药品审评报告中所反映出的历年药品审评审批制度改革的重点,以及药品注册申请存在的主要问题进行的概括性梳理。从中不难一览CDE朝高效、优质的药品审评制度不断进步的改革决心,以及近年工作重心的变更脉络。此外,历年药品审评报告中所总结的主要问题,也为业界同仁今后的药品注册申请工作提供有益参考。在中篇和下篇中,我们将继续结合历年制度改革重点,分别从审评审批时限管理、化学药品注册分类变化与申请变化、仿制药一致性评价、生物制品注册发展、儿童用药与罕见病药物审评审批工作这五大主题出发,进一步解读近年来我国药品审评审批的改革重点和制度发展。欢迎持续关注。
Originally Published 15 October 2022
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]

