
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- in China
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- in China
- within Food, Drugs, Healthcare, Life Sciences, Technology and International Law topic(s)
在《中国年度药品审评报告解读(2015-2021)——回顾中国药品审评审批制度改革成果(上篇)》中,我们对历年年度药品审评报告中提及的药品审评审批制度改革重点内容进行了概括性提炼,本文的中篇和下篇将结合上篇中提到的历年制度改革重点,进一步从审评审批时限管理、化学药品注册分类变化与申请变化、仿制药一致性评价、生物制品注册发展、儿童用药与罕见病药物审评审批工作五大主题出发,深入解读近七年药品审评审批制度的发展。在本篇中,我们主要将围绕前三大主题进行探讨。
主题一:审评审批时限管理
(一) 药品审评积压现象及成因
在很长的一段时间内,药品审评积压(Drug Lag)是我国药监部门面临的严重问题。《2015年度药品审评报告》中也明确指出"审评任务大量积压已成为实现科学监管和行业良性发展的巨大障碍"。在2015年高峰时期,国家药品监督管理局药品审评中心(下称 CDE)的审评任务积压达到了22,000多个。因此,如何解决药品审评积压问题,成为了药监部门面临的艰巨任务。
药品审评积压问题是由多重原因造成的:首先,审评工作量与审评人员规模的失衡导致积压问题难以解决。2000年后,药品审评审批权由中央上收,原本分散在各省的审评量都集中到了中央。审评工作量激增,使得有限的审评人力资源捉襟见肘。其次,冗长的监管审查时间和不断延迟的审评审批时限也是造成积压问题的重要原因。据研究统计,2013年至2015年间,我国创新药物临床试验申请审评审批的平均延迟时间为14个月。 1最后,企业申报质量不高,低水平重复现象严重,申报资料不完整、不真实、不规范等问题严重,进一步导致药品审评积压。
(二)主要应对措施
《国务院关于改革药品医疗器械审评审批制度的意见》(国发〔2015〕44号)(下称" 44号文")将解决注册申请积压作为药品医疗器械审评审批制度改革的主要目标之一,并提出了"争取在2016年底前消化完积压存量,尽快实现注册申请和审评数量年度进出平衡,2018年实现按规定时限审批"的工作目标。
为了解决药品审评积压问题,近年来我国主要采取了包括但不限于以下几种措施:
第一,开展药物临床试验数据自查核查工作。如前所述,企业申报资料不真实、不规范问题严重是导致审评积压的原因之一。为此,2015年7月22日,原国家食品药品监督管理总局发布《关于开展药物临床试验数据自查核查工作的公告》,组织对已申报生产或进口的待审药品开展临床试验数据核查工作。其中要求1622个临床试验项目自查,如有问题主动撤回。如果在规定时间没有提交报告或撤回,总局将进行飞行检查,一旦查出问题,相关申请人、临床试验机构和相关责任人员将面临严重的行政处罚后果。截至2015年12月31日,申请人主动撤回药品注册申请达1009个,占比62.2%,涉及药品企业数百家。其后,随着《药物临床试验数据核查工作程序(暂行)》《药品管理法》《药品注册管理办法》《药品注册核查检验启动工作程序》《药品注册核查工作程序(试行)》《药品注册核查要点与判定原则(药物临床试验)(试行)》等规定的陆续出台,药物临床试验数据的自查核查工作从一次突如其来的核查风暴逐步演进为成熟的长效监管机制。药物临床试验数据自查核查工作的开展,一方面减少了低质量药品注册申请的数量,另一方面提升了药品注册申请的整体质量,有效促进了药品审评积压问题的解决。
第二,确立临床试验的默示许可制度。如前所述,审评审批时限的延迟尤其是临床试验申请审批时限的延迟是造成积压问题的原因之一。对此,2017年,中共中央办公厅、国务院办公厅发布的《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》(厅字〔2017〕42号)(下称" 42号文")中提到要优化临床试验审批程序,受理临床试验申请后一定期限内,食品药品监管部门未给出否定或质疑意见即视为同意,注册申请人可按照提交的方案开展临床试验。此后,2019年修订的《药品管理法》第19条正式确立了我国临床试验申请的默示许可制度,将临床试验审评审批的时限大幅缩短至60个工作日之内,进而大大推动了我国药品临床试验审评审批的进程。
第三,设置药品加快上市注册程序。2020年修订的《药品注册管理办法》第四章分别对突破性治疗药物程序、附条件批准程序、优先审评审批程序和特别审批程序进行了原则性的规定。后续《突破性治疗药物审评工作程序(试行)》《药品附条件批准上市申请审评审批工作程序(试行)》《药品上市许可优先审评审批工作程序(试行)》等文件的陆续出台进一步推动了药品加快上市注册程序的成熟落地。此外,《药品注册管理办法》第96条明确规定了优先审评审批程序的审评时限为130个工作日,临床急需境外已上市罕见病用药优先审评审批程序的审评时限为70个工作日,在加快审评审批时限的同时提升了审批时限的可预见性。
第四,扩增审评力量。从《2019年度药品审评报告》指出"大规模招聘人员、借调省局人员等措施多渠道扩增审评力量"到《2021年度药品审评报告》指出"持续加强审评队伍和能力建设""积极协调增加人员编制,立足审评需要做好人才引进工作,充实专业审评力量",推进人才队伍建设始终是CDE近年来的工作重点。CDE通过招聘人员、借调省局人员、协调编制等方式,积极扩增审评力量,为解决审评积压问题作出重要努力。
(三)政策效果
根据历年年度药品审评报告中的数据显示,2015年至2018年间,我国药品审评积压问题已经逐步得以解决。2015年,审评任务积压由高峰时的22,000多个降至年底的不到17,000个;2016年,排队等待审评的注册申请下降至近8200件;2017年,排队等待审评的注册申请下降至4000件,基本完成了44号文确定的解决药品注册申请积压的工作目标;2018年,排队等待审评的注册申请降至3440件,进一步巩固了44号文要求解决注册申请积压的成效。
《2019年度药品审评报告》中指出,2019年,药品审评申请积压基本得以解决,CDE的工作重点已经由解决药品注册申请积压逐渐过渡为提升药品注册申请的按时限审评审批率。从CDE公布的每一年的数据来看,2019年至2021年间,我国药品审评全年整体按时限审结率逐年提升。2019年,CDE实现了中药、化学药、生物制品各类注册申请按时限审评审批率超过90%;2020年,全年审结注册申请任务整体按时限完成率为94.48%;2021年,按时限审结率达到了98.93%。
主题二:化学药品注册申请要求
的变化
(一)化学药品注册分类变化
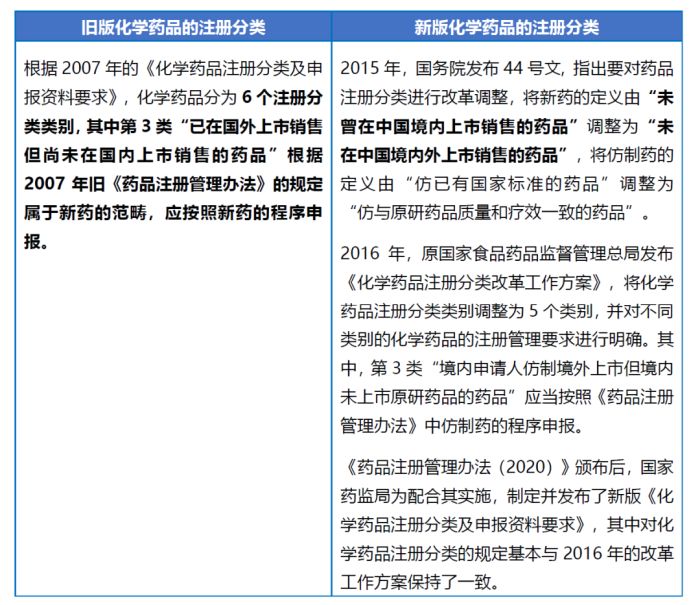
我国于2016年对化学药品的注册分类进行了调整,其中对新药和仿制药的定义发生了改变,使新药的范围缩小、仿制药的范围扩大。与2007年的注册分类要求相比,目前,我国对新药的定义从"中国新"转变为"全球新",即新药必须在境内外均未上市,并进一步将新药分为1类新药(创新药)和2类新药(改良型新药)。过去归属于新药范畴的"境内申请人仿制境外上市但境内未上市原研药品的药品"现被认定属于仿制药范畴。注册分类的变化情况如下表所示:
(二)申报资料要求与国际接轨
对比2007年与2020年的《化学药品注册分类及申报资料要求》,我国在化学药品注册申报资料的要求上也发生了变化,主要体现在2020年《化学药品注册分类及申报资料要求》要求参照国际人用药品注册技术协调会(ICH)的指导原则来明确申报资料要求。
ICH是在全球具有广泛影响力的人用药品注册技术协调机构。我国于2017年正式加入ICH,国家药监部门于2018年成为ICH管理委员会成员。为推动药品注册技术标准与国际接轨,原国家食品药品监管总局于2018年1月发布《关于使用人用药品注册技术国际协调会二级指导原则的公告》,明确自2018月2月1日起,化学药品注册分类1类和5.1类、治疗用生物制品1类及预防用生物制品1类注册申请适用于《M4:人用药物注册申请通用技术文档(CTD)》。
2020年国家药监局制定的新版《化学药品注册分类及申报资料要求》中要求,申请人提出药物临床试验、药品上市注册及化学原料药申请,应按照国家药品监管部门公布的相关技术指导原则的有关要求开展研究,并按照现行版《M4:人用药物注册申请通用技术文档(CTD)》格式编号及项目顺序整理并提交申报资料。
(三)分类和申报资料要求的变化对化学药注册申请的影响
01 整体影响
首先,新药注册的研发创新水平门槛提升。在新的化学药品注册分类下,我国对新药的定义从"中国新"转变为"全球新",使得新药的范围缩小、对企业的研发能力要求提升。同时,新化学药品注册分类中将新药进一步分为1类创新药和2类改良型新药。其中,对改良型新药而言,要求其相较于被改良的药品,要具有"明显临床优势",这将有效遏制仅更改剂型、仅更改给药途径等低创新水平的产品注册成为新药的可能性。
其次,仿制药注册面临更严格的监管要求。新版化学药品注册分类对仿制药的定义更加准确、要求更加严格,强调仿制药与参比制剂在质量和疗效方面的一致性。在旧分类下被列为新药的第3类境外已上市但境内未上市的药品,在新分类下被归为仿制药的范畴,相应的新药监测期被取消,但对其审评的要求却不降反增,监管要求其与原研药品开展一致性评价。因此,企业仿制第3类境外已上市但境内未上市的药品,将受到更加严格的高标准要求。
02 对第3类化学药注册申请的影响
第3类化学药申报资料要求的变化导致其注册申请结果受到影响。《2021年度药品审评报告》中指出,"因缺乏境内有效性、安全性临床数据而未获批准的化学药注册分类第3类上市注册申请数量较往年明显增加",这主要与"现行《药品注册管理办法》实施后化学药注册分类第3类上市注册申请审评结论管理要求的调整有关"。
由此可见,对于调整后的第3类化学药注册申请要求,在2021年业界仍处于消化接受的过程之中。值得注意的是,第3类化学药品上市注册申请并不完全豁免境内临床试验要求。2020年《境外已上市境内未上市药品临床技术要求》中规定了第3类药品上市注册的临床试验要求。其中明确,基于种族敏感性分析、原研药品完整临床试验数据可得性、仿制药制剂学等因素的考虑,境外已上市境内未上市药品仍需开展临床试验,以评估中国患者用药的安全性和有效性。
主题三:仿制药一致性评价制度发展
(一)一致性评价制度的建立与完善
2015年国务院44号文提出,要将仿制药的定义由"仿已有国家标准的药品"调整为"仿与原研药品质量和疗效一致的药品",强调仿制药要与原研药品在质量和疗效方面要能够达到一致的水平。建立与完善仿制药一致性评价制度,有助于提升我国仿制药的质量、促进我国制药产业的发展,切实保障用药安全。
01 政策依据
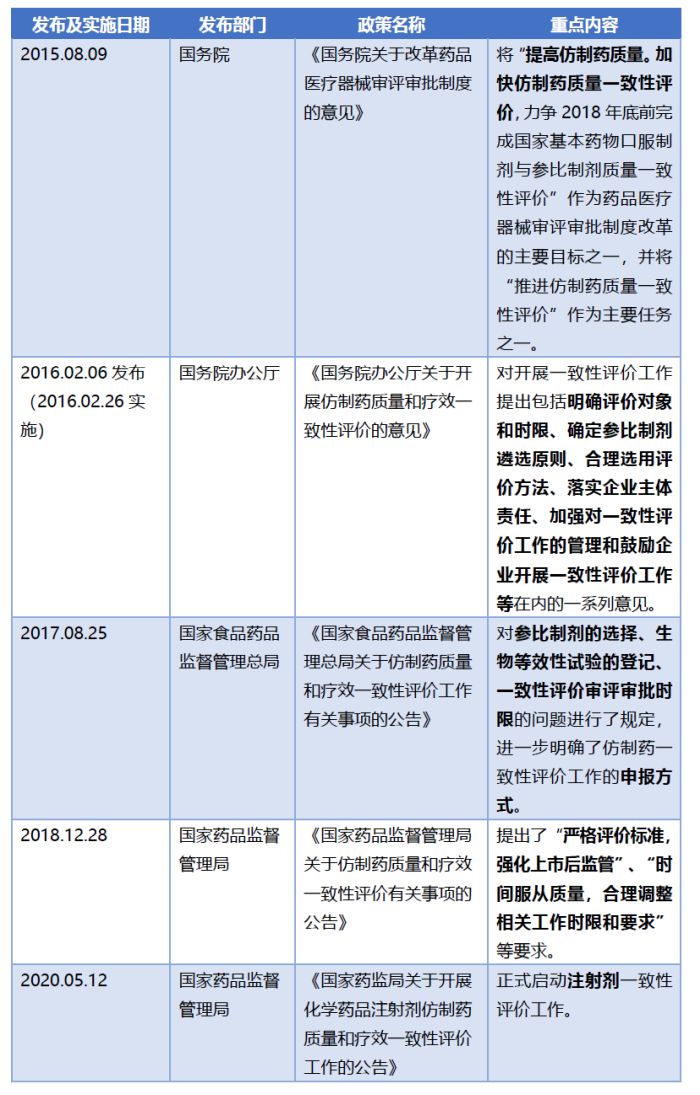
2015年至今,我国发布了一系列政策文件,以推进仿制药一致性评价制度的建立与完善。
02 仿制药一致性评价工作的开展情况
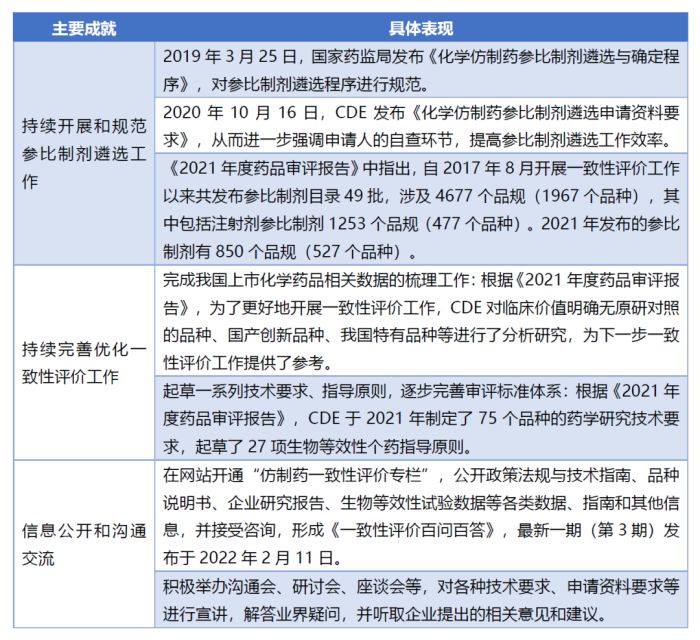
CDE从2017年8月开始正式承担仿制药质量和疗效一致性评价整体工作。自2017年以来,CDE每年的工作重点内容都包括了大力开展和推进仿制药一致性评价工作。具体而言,近年来CDE在推进仿制药一致性评价工作方面取得的主要成就如下表所示:
03 289基药目录一致性评价工作延期
如前所述,我国将仿制药的定义由"仿已有国家标准的药品"调整为"仿与原研药品质量和疗效一致的药品",这一改变表明监管机关对于仿制药的审评要求变得更加严格,强调仿制药与参比制剂在质量和疗效方面的一致性。在此之前,经我国批准上市的仿制药并没有与原研药品一致性评价的强制要求,使得部分仿制药在质量和疗效上与原研药品存在一定的差距。因此,为了推进在化学药品新注册分类实施前已经批准上市的仿制药开展一致性评价工作,国务院办公厅于2016年2月发布了《关于开展仿制药质量和疗效一致性评价的意见》规定,国家基本药物目录(2012年版)中2007年10月1日前批准上市的化学药品仿制药口服固体制剂(常称" 289基药目录"),应在2018年底前完成一致性评价,其中需开展临床有效性试验和存在特殊情形的品种,应在2021年底前完成一致性评价;逾期未完成的,不予再注册。
然而,据GBI SOURCE数据库显示,截至2018年12月28日,289基药目录中仅有62个品规通过或视同通过仿制药一致性评价,涉及32个品种。 2这样的现实与上述目标的差距甚远。于是,当时有不少业内人士提出,我国的很多仿制药企业经营规模较小、经济实力不佳,通过一致性评价所需的成本对于这些中小企业而言是不小的负担,考虑到这些企业的实际情况,希望在政策层面能够放宽相应品种完成一致性评价的期限。
2018年12月28日,国家药品监督管理局发布《关于仿制药质量和疗效一致性评价有关事项的公告》,提出基于"时间服从质量"的考虑,对相关工作时限和要求进行了合理的调整:对已经纳入国家基本药物目录的仿制药品种,不再统一设置一致性评价时限要求;化学药品新注册分类实施前批准上市的含基本药物品种在内的仿制药,原则上应自首家品种通过一致性评价后3年内完成一致性评价,逾期未完成的,企业经评估认为属于临床必需、市场短缺品种的,也可由所在地省级药品监管部门经申请认定后,予以适当延期。考虑到基本药物对公众临床用药的重要性,2018年国家药监局对289基药目录一致性评价工作的延期安排是立足于我国实际情况做出的科学与合理调整。
(二)一致性评价品种趋于丰富化
根据近年的年度药品审评报告显示,在2017至2021年间,CDE批准的一致性评价品种持续上升(通过一致性评价的品种按照通用名统计)。2017年,CDE完成首批52件一致性评价申请的审评工作,其中通过一致性评价药品共13个品种(17个品规);而到了2021年,通过一致性评价的品种达到了331个品种,实现了明显的增长。一致性评价品种的丰富,一方面,在确保药品质量和疗效的有效性的情况下,使得公众用仿制药的选择范围不断扩大且用药成本得到有效的降低;另一方面,也为医药企业的竞争发展和医保集中招标采购工作的开展注入生机。
(三)一致性评价申请按时限审结率总体较高
原国家食品药品监督管理总局于2017年发布的《关于仿制药质量和疗效一致性评价工作有关事项的公告》中对一致性评价的审评时限要求进行了明确:国家食品药品监督管理总局行政事项受理服务和投诉举报中心应在签收资料5个工作日内,对申报资料进行形式审查并决定受理与否;受理后,CDE应对企业申报资料进行立卷审查,并对符合要求的于45个工作日内完成立卷;CDE一般应当在受理后120个工作日内完成审评工作;经审评认为需申请人补充资料的,申请人应在4个月内一次性完成补充资料。如需申请人补充资料,补充资料回复后的技术审评时限为40个工作日,行政审批时限为20个工作日。
CDE非常重视仿制药一致性评价工作的开展,因此集中相关审评资源,优先审评仿制药的一致性评价申请。根据2019年底的统计数据,一致性评价的按时限审结率总体在90%以上,审评时限的中位值大约为124个工作日(包括各轮审评时限总和)。 3《2021年度药品审评报告》中显示,2021年CDE一致性评价注册申请按时限审结率达到了98.80%。
以上为我们针对审评审批时限管理、化学药品注册分类变化与申请变化以及仿制药一致性评价三大主题的讨论,我们将在下篇文章中继续讨论生物制品注册发展和儿童用药与罕见病药物审评审批工作这两大主题,欢迎持续关注。
Footnotes
1. Zhou Q, Chen XY, Yang ZM, Wu YL. The changing landscape of clinical trial approval processes in China [J]. Nat Rev Clin Oncol,2017,14(9):577-583.
2. 张雪.药品一致性评价:取消大限并非无限延期[N].上海证券报,2019-1-3
3. 陈新,吴倩,温宝书.化学仿制药质量和疗效一致性评价工作进展及展望[J].中国新药杂志,2020,29(19):2185-2189.
Originally Published 16 October 2022
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]

