
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- within International Law, Technology and Corporate/Commercial Law topic(s)
一、背景
近日,网传国家药品监督管理局药品注册管理司于7月20日发布了《关于加强药物临床试验机构日常监管的函》(" 网函")1,其中提及国家药监局核查中心联合相关省局在组织开展药品注册现场核查中,发现13家临床试验机构在临床试验数据完整可靠方面存在问题。核查中心已将核查发现的问题告知被检查单位,并建议整改。核查中心将现场核查发现的问题通报省局,指示省局监督相关机构在6个月内完成整改,整改到位并经省局确认后,上述被检查单位方可开展新的药物临床试验。各省局可视情况运用告诫、约谈、警告等措施依法依规处理。
由于尚未能在官方渠道查到该文件,该函的真实性尚未得到确认,但本文旨在从法规及行业合规实践的角度就相关药品注册核查问题予以提示。
二、药品注册核查一贯为药监
部门执法重点
我国药监部门一贯重视药品注册流程中对于临床试验数据真实性与合规性的核查。即便过去数年,医药圈对于"722风暴"大抵都记忆犹新。自2015年7月22日《关于开展药物临床试验数据自查核查的公告》一文发布以来,国家药监局以破釜沉舟的革新气势开启了药物临床试验数据的大规模自查核查工作,严肃处置临床试验数据在过往行业实践与监管过程中存在的沉疴弊病,很大程度上净化了医药研发的生态环境、促进企业的理念的转变。依据《药物临床试验数据核查阶段性报告(2015年7月-2017年6月)》,两年内,国家药监局共要求2033个药品注册申请开展临床试验数据"自查核查"。核查期间,1316个药品注册申请由申请人主动撤回;313个药品注册申请受到核查中心的现场核查,其中38个注册申请的临床试验数据经核查涉嫌数据造假进而面临进一步的处理。
"722风暴"后,我国药监部门持续高度重视药物临床试验数据的合规问题,不断完善临床试验数据核查体系,所出台的诸多法规文件皆对临床试验数据的核查工作作出相应的规范。由此,临床试验数据核查工作从一次突如其来的监管风暴逐步演进为成熟完善的长效监管机制。
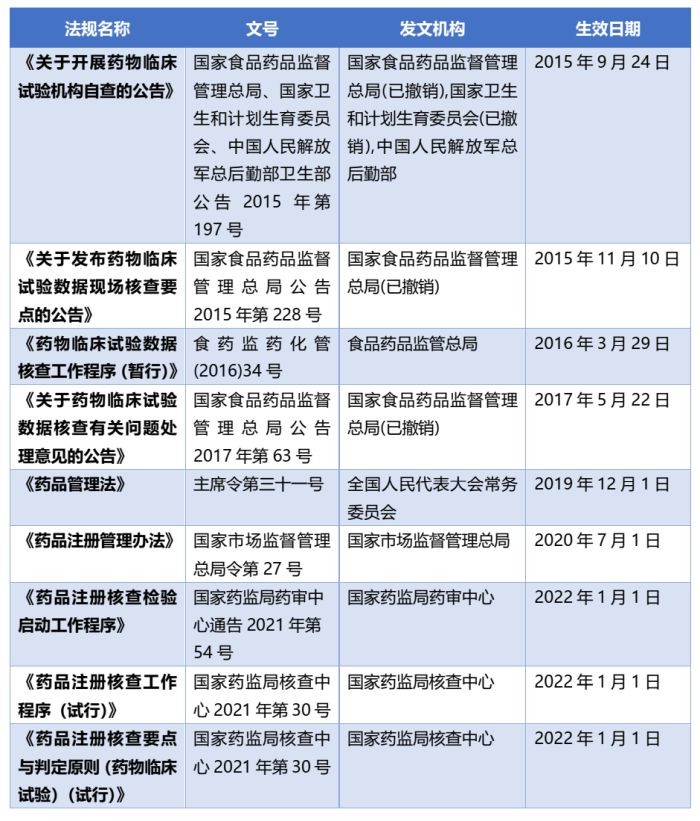
此外,根据《2018年度药品审评报告》,创立基于审评需要和风险控制的检查检验原则与标准,探索以审评为主、检查检验为支撑的工作机制和程序是药品审评中心2018年的主要工作方向。根据《2020年度药品审评报告》,药审中心正积极转变药品注册核查理念,将注册现场核查启动工作模式由基于审评需要调整为基于风险启动。从监管理念到工作模式,近年来我国药监部门积极推动药品注册核查工作走向成熟化与规范化。
从监管实践看,药物临床试验的注册核查工作始终是我国药监部门执法的重点关注对象。自2019年至2021年,药审中心启动临床试验数据核查任务分别为446个、439个、383个。此次网函发布前,实践中亦存在类似的监督检查案例:根据国家局的临床试验机构登记网站公示信息,国家局曾于2021年7月8日向河南省药监局发函,要求河南省药监局监督当地某医院对现场核查中发现的知情同意书签署等方面的问题进行整改,该机构在整改到位并经所在地省局确认后方可开展新的药物临床试验项目。
三、实施方案内容解析
若网函内容属实,则其体现的核查内容及监管动向不无参考意义,本文将稍加分析。网函提及的"药品注册现场核查"并非药品注册流程中的必经检查项目,而是由药品审评中心基于根据药物创新程度、药物研究机构既往接受核查情况等,基于风险决定是否开展药品注册现场核查 2。具体而言,研发生产主体合规因素,包括生产制造的相关单位和机构既往接受核查的情况,都会成为是否启动药品注册核查考虑的风险因素 3。由此可见,研发生产主体及其他相关单位维护良好的合规记录,有助于降低药品注册中的合规风险,避免药品注册期限因受到额外审查而延长。
值得注意的是,网函并非直接面向13家临床试验机构作出的行政处罚,而是国家药监局药品注册司向省局通报核查发现的问题。依《药品注册核查工作程序(试行)》第41条规定,根据报送的现场核查问题及相关材料,省局药品监督管理部门依日常监管职责对被核查单位的现场核查发现问题整改情况进行审核确认,必要时进行跟踪检查,并将审核结果及时告知药品审评中心。据此,收函的十省药品监督管理局后续将监督相关单位进行整改。目前该函提及"可视情况运用告诫、约谈、警告等措施依法依规处理",其中"告诫"、"约谈"也并非行政处罚手段。但需要注意的是,若后续仍存在整改不到位、临床试验数据完整可靠方面存在问题的情况,不排除对被检查单位及相关责任人另行采取处罚措施。例如,根据《药品管理法》第123条规定,提供虚假的证明、数据、资料、样品或者采取其他手段骗取临床试验许可或者药品注册等许可的,撤销相关许可,十年内不受理其相应申请,并处五十万元以上五百万元以下的罚款;情节严重的,对法定代表人、主要负责人、直接负责的主管人员和其他责任人员,处二万元以上二十万元以下的罚款,十年内禁止从事药品生产经营活动,并可以由公安机关处五日以上十五日以下的拘留。
四、合规启示
药品注册核查,体现了药监部门及时发现问题,高效沟通解决问题,保护受试者安全权益的决心;对于进一步提高相关被检查单位的临床研究合规能力,加强相关省份药物监督日常管理,提升属地整体临床试验管理水平具有重要意义。
申办者是保证临床数据质量的最终责任人,也是开展药物临床试验的委托人和受益人。相应地,当临床试验数据完整可靠性发生问题时,申办者也蒙受最大的损失。因此,建议申办者可在以下方面加强管理:
(一) 明确认识自身主体责任
a) 申办方应充分认识其主体责任,必须保证药品注册申请中临床试验数据符合真实、完整和规范的要求,并对数据管理过程的合规性负有监督责任;
b) 当工作涉及外包时,申办者应对合同研究组织(CRO)、现场管理组织(SMO)工作过程和成果的合规性以及数据的质量进行监督。
(二) 加强对临床试验机构和研究者的监督,在临床试验协议中明确以下内容
a) 临床试验机构和研究者应保证临床试验数据的真实、完整和规范;
b) 临床试验机构和研究者应保证以病例报告表(CRF)等形式报告给申办者的数据准确、完整与及时;
c) 临床试验机构和研究者应保证CRF上的数据来自于受试者病历上的源数据,并应当对其中的任何不同予以合理解释;
d) 临床试验机构和研究者应及时响应数据管理员、监察员的问题和质疑,并及时清理发现的任何问题;
e) 临床试验机构和研究者应保证其具备相应的资质,且进行具体活动的相应人员也具备资质;
f) 临床试验机构和研究者应保证严格遵守研究方案。
(三) 加强对合同研究组织(CRO)、现场管理组织(SMO)的监督,在服务协议中明确以下内容
a) CRO和SMO应当保证服务过程和成果的质量,并实施相应的质量保证和质量控制措施;
b) CRO和SMO应当对相关员工进行相关的培训,以保证其所提供的服务符合申办者和法律法规提出的质量标准要求;
c) 申办者有权对CRO和SMO进行的活动进行及时有效的管理、沟通和核查,以确保其遵守共同商定的流程要求。
(四) 加强临床试验方案设计能力,完善临床研究管理体系:
a) 申办者应建立包括完善的临床开发职能的临床研究管理体系,覆盖负责制定药物开发策略、设计并开展临床试验、数据质量控制、不良反应监测、分析数据结果并申报上市等临床研究全流程各个环节;
b) 申办者应制定标准操作流程(SOP),根据自身特点和内部职能设定,制定符合GCP要求的SOP体系,并确保严格执行;
c) 申办者可以通过数据信息化系统,系统性地增加临床试验管理的质量和效率。
同时,国家药监局注册司在该函中也指示省局加强日常监管,监督持续实施GCP,确保临床试验开展的合规性和临床数据质量的可靠性,保护受试者安全权益。可见,未来保障人民群众用药安全,药品注册核查将继续成为工作重点。相关药品的申办方与临床试验机构应当引起足够重视,并且进一步加强临床试验中对合同研究组织、临床试验现场管理组织等第三方机构的监管,建立完善的临床研究管理体系。
Footnotes
1. 根据网络公开信息,该函公开属性为依申请公开。
2.《药品注册管理办法》第46条。
3.《药品注册核查检验启动工作程序》第9条。
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]

