

- within Food, Drugs, Healthcare and Life Sciences topic(s)
- in India
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- in India
- within Food, Drugs, Healthcare, Life Sciences, Employment and HR and Corporate/Commercial Law topic(s)
- with Inhouse Counsel
- with readers working within the Business & Consumer Services, Chemicals and Retail & Leisure industries
Introduction
A clinical trial1 for drugs and medical devices involves systematic studies with human subjects to evaluate their clinical and pharmacological effects, including adverse effects, aiming to determine their safety, efficacy, or tolerance. In India, the term 'clinical trial' is used for investigation of drugs before regulatory approvals, while the corresponding term for medical devices is 'clinical investigation'. The market for clinical trials and investigation in India is expected to grow at CAGR of 8.2% from 2022 to 20302. With a diverse genetic pool, abundance of technical resources and cost-effectiveness, India is well-positioned to become a global hub for clinical trials and investigation. To regulate this growth, India has formulated a legal framework to ensure the safety of participants, and the integrity of the data collected, while at the same time providing certainty and regulatory clarity to the industry.
Regulatory Framework
Regulatory Authorities
The Central Drugs Standard Control Organization (CDSCO) is the apex regulatory body overseeing the pharmaceutical industry including the approvals required for clinical trials and clinical investigations. The Drugs Controller General of India (DCGI), the head of CDSCO, is responsible for granting permission for clinical trials and investigations and regulating the importation and sale of drugs and medical devices in India. In addition, the Central Licensing Authority (CLA) and Ethics Committees have been established to provide or deny approvals for trials and investigations.
The Indian Council of Medical Research (ICMR) also plays a major role by hosting the Clinical Trial Registry of India (CTRI) portal which is an online public record system for registration of ethics committees and clinical trials being conducted in India. All clinical trials, including global clinical trials3, in the country are required to be registered with the CTRI as a prerequisite for enrolling subjects for the trial.
All the above-mentioned regulatory bodies come under the ambit of one central government ministry, i.e., the Ministry of Health and Family Welfare (MoHFW). The MoHFW by itself also controls certain aspects of pharmaceutical sector. One major way in which it does that is by releasing a list of medicines called the National List of Essential Medicines (NLEM) that are the most essential drugs for the general population of the country.
In addition, the Department of Pharmaceuticals (DoP) also governs certain aspects of the pharmaceutical industry. A major role that DoP plays in the Indian pharmaceutical sector is through the National Pharmaceutical Pricing Authority (NPPA) by fixing ceiling prices for certain drugs which it deems to be critical for the general population. The legal instrument by which the prices are fixed is the Drugs (Price Control) Order, 2013 (DPCO). The DPCO takes reference from the NLEM and fixes the maximum price for the drugs listed in the NLEM.
Legal Framework
The legal system in India has a tiered structure where the government formulates a principal legislation, called an 'Act', to cover a subject broadly. The Act consists of subject and aspects of it that it aims to regulate, such as regulation of a specific business, requiring license to operate a business, approval to import goods, etc. The procedural aspects of the subject covered in the Act are then provided in the subordinate legislations, i.e., 'Rules', 'Regulations' or 'Guidelines' which are framed by concerned regulatory bodies as per the powers accorded to them under the principal Act. The subordinate legislations specify the procedures and other supplementary details on the subject matter of the Act, such as procedure to apply for a license, conditions to fulfil for qualifying for import license, etc.
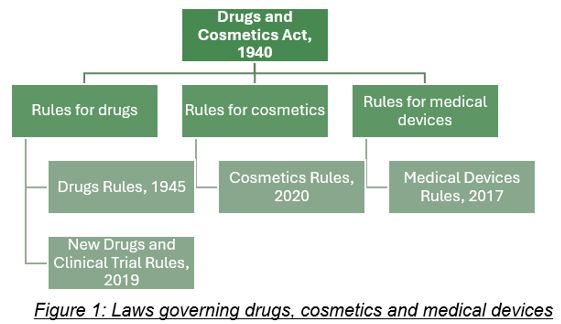
The principal Act that regulates the pharmaceutical industry (a term which generally includes drugs and medical devices both) in India is the Drugs and Cosmetics Act, 1940 (DC Act). The DC Act within its ambit regulates drugs, cosmetics, medical devices (including softwares as medical device (SAMD)) and their approval, manufacturing, import, licensing, quality standards and distribution and sale. The major subordinate legislations under the DC Act are the Drugs Rules, 1940 (Drugs Rules), Cosmetics Rules, 2020 (Cosmetics Rules) and the Medical Devices Rules, 2017 (MD Rules).
However, it should be noted that the definition of 'drug' under the DC Act includes medical devices as well. This leads to an ambiguity in law since 'medical device' is separately defined and regulated under the MD Rules. This ambiguity is practically dealt with by the medical devices industry in two ways: (i) by complying with both the subordinate legislation, i.e., Drugs Rules and the MD Rules as far as practicable; and (ii) where the DC Act and subordinate legislations are clear is their applicability, by complying with only the provisions which are applicable to medical devices.
Laws for clinical trials and investigations
For the clinical trial and approval of drugs, the New Drugs and Clinical Trial Rules, 2019 (NDCT Rules) was enacted. The NDCT Rules, in its ambit, covers clinical trials, academic clinical trials4, bioavailability study5, and bioequivalence study6.
On the other hand, the MD Rules provide regulatory framework for medical devices, their use, import, sale, and clinical investigation. The MD Rules regulate the academic clinical study7, clinical investigation8 and clinical performance evaluation9 of medical devices. It should be noted that NDCT Rules use the term 'clinical trial' for the investigation of drugs before regulatory approval, while the MD Rules use the term 'clinical investigation' for medical devices.
In addition to these legislations, the ICMR has also issued a set of guidelines called the National Ethical Guidelines for Biomedical and Health Research involving Human Participants, 2017 (ICMR Guidelines) to ensure ethical standards and the protection of participants in research studies conducted in India.
Stages of Clinical Trial of Drugs
Four phases of drug trial: The NDCT Rules lays down a 4-phased approach to clinical trials facilitating a systematic progression from initial safety testing in small groups to broader assessments of efficacy and safety in larger patient populations. Phase I involves a small group of healthy volunteers or patients to assess safety, pharmacokinetics, and pharmacodynamics. Phase II evaluates the drug's effectiveness, short-term side effects, and risks in a homogeneous patient population, and explores study endpoints, therapeutic regimens, and target populations. Phase III enrols a larger population to confirm efficacy, monitor side effects, and compare the drug with standard treatments, providing a basis for marketing approval. Phase IV occurs post-marketing to monitor long-term safety and effectiveness in real-world settings, including studies on drug interactions, dose responses, and specific indications.
Waiver for drug trials: For drugs developed outside India or trials conducted outside India, the CLA may waive Phase I trial in India if a request is submitted to it along with the Phase I data from the study conducted outside India. If the CLA is not satisfied, it may require the trial to begin from Phase I in India. Further, in certain life threatening or serious disease conditions or rare diseases or for unmet medical needs in India, the CLA may relax, abbreviate, omit or defer the requirement of non-clinical and clinical data on case- to-case basis. The CLA is also empowered to issue a list of countries for considering waiver of local clinical trials for approval of new drugs. As of now, certain categories of drugs have been waived clinical trial in India if they have regulatory approval in Japan, the United States, the United Kingdom, Australia, Canada and the European Union.
Stages of Clinical investigation of Medical Devices
Three stages of medical device investigation: The MD Rules specifies a three staged investigation method which includes pilot clinical investigation, pivotal clinical investigation and post marketing clinical investigation. Pilot clinical investigations are exploratory studies conducted with a small number of patients to gather essential information about a medical device's performance and safety before larger pivotal studies. Objectives include assessing feasibility, eligibility criteria, potential harm, device mechanisms, and logistics for future studies. Pivotal clinical investigations are definitive studies that gather evidence to support the safety and effectiveness of the device in a larger patient population, and for devices without a predicate10 but approved elsewhere, these studies generate evidence specific to Indian patients. Post marketing clinical investigations, conducted after marketing approval, optimize the device's intended use and may include additional safety studies, drug-device interaction evaluations, and investigations to support use under approved indications.
Waiver of clinical investigation: For clinical investigation of medical devices, the CLA may, in public interest, abbreviate, defer, or waive the requirement of clinical data for conducting clinical investigation before granting permission to conduct clinical investigation. Further, for medical devices that are substantially equivalent to a predicate device, the CLA may approve the said device for marketing without requiring clinical investigation. As of now, if a medical device is approved by the regulatory authorities of Japan, the United States, the United Kingdom, Australia or Canada and the said device has been marketed for at least two years in that country, the clinical investigation may be waived off based on case-to-case analysis.
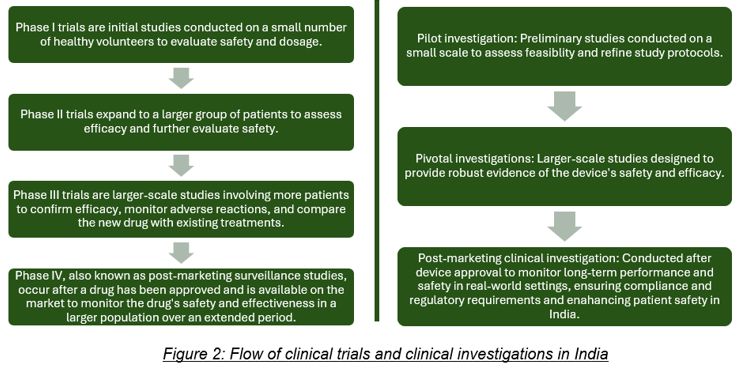
In brief, following is the flow of clinical trial and investigation:
Each phase/stage plays a role in progressively gathering data to determine the drug's or the medical device's suitability for regulatory approval and clinical use.
Common elements between clinical trials and investigations: Even though the approval of new drugs and medical devices are covered under different rules, there are various overlapping procedures which are common for both. Some of the common procedures include formulation of trial / investigation protocol, approval of such protocol being the first step in the trial / investigation, approval from an ethics committee, stage wise development of the trial / investigation, etc.
For both, i.e., clinical trials of drugs and clinical investigation of medical devices, registration with the CTRI is relevant. In addition, approvals from the Ethics Committee and the CLA are equally important to conduct a clinical trial or clinical investigation in India. Ethics Committees are a statutory body registered with the CLA, tasked with the responsibility to review and accord its approval to clinical trials and clinical investigations.
Approach to clinical trials and investigations
Practically, a common way that companies adopt to conduct clinical trials and investigations in India is through Contract Research Organizations (CRO). CROs are usually contracted to assist in study design and planning, ensuring protocols meet regulatory requirements and developing detailed project plans. CROs can also handle regulatory submissions, compliance with local and international standards and can also conduct quality assurance audits and manage post-marketing surveillance to monitor long-term safety and effectiveness. Through these services, CROs enable sponsors11 to focus on their core competencies, ensuring high-quality data and successful trial outcomes.
Future Directions
Looking ahead, India is poised to strengthen its position as a global hub for clinical trials by leveraging emerging technologies and innovations in healthcare. The integration of artificial intelligence (Al) and big data analytics is expected to revolutionize clinical trial design, patient recruitment, and data analysis, thereby enhancing the efficiency and cost-effectiveness of trials.
India has, in recent times, taken steps to make effective advancement in this sector by streamlining the regulatory framework to accelerate the process of clinical trials and investigations in the country. The country's health ministry has established a screening committee called the Health Ministry's Screening Committee (HMSC) for specifically taking decisions on international research proposals in the field of health research requiring foreign collaboration and/or assistance from foreign funding agencies. Further, the online registry of clinical trials, i.e., the CTRI, serves as a transparent public record system that allows users to register clinical trials, search for trials, and download trial registration sets and publications.
These advancements in digital health solutions will not only improve patient recruitment and retention but will also enable real-time monitoring of patient outcomes, ensuring timely intervention and data collection.
Footnotes
1 Rule 2(1 )(j), NDCT Rules: "clinical trial" in relation to a new drug or investigational new drug means any systematic study of such new drug or investigational new drug in human subjects to generate data for discovering or verifying its: (i) clinical or; (ii) pharmacological including pharmacodynamics, pharmacokinetics, or; (Hi) adverse effects, with the objective of determining the safety, efficacy or tolerance of such drug or investigational new drug.
2 Business Wire, The Indian Clinical Trials Market Size, Share and Trend Analysis: https://www.businesswire.com/news/home/20220308006146/en/Indian-Clinical-Trials-Market-Size-Share-Trends-Analysis-2022-2030---ResearchAndMarkets.com
3 Rule 2 (1)(q), NDCT Rules: "global clinical trial" means any clinical trial which is conducted as part of a clinical development of a drug in more than one country.
4 Rule 2(1 )(a), NDCT Rules: "academic clinical trial" means a clinical trial of a drug already approved for a certain claim and initiated by any investigator, academic or research institution fora new indication or new route of administration or new dose or new dosage form, where the results of such a trial are intended to be used only for academic or research purposes and not for seeking approval of the Central Licensing Authority or regulatory authority of any country for marketing or commercial purpose.
5 Rule 2(1 )(e), NDCT Rules: "bioavailability study" means a study to assess the rate and extent to which the drug is absorbed from a pharmaceutical formulation and becomes available in the systemic circulation or availability of the drug at the site of action.
6 Rule 2(1 )(f), NDCT Rules: "bioequivalence study" means a study to establish the absence of a statistically significant difference in the rate and extent of absorption of an active ingredient from a pharmaceutical formulation in comparison to the reference formulation having the same active ingredient when administered in the same molar dose under similar conditions.
7 Rule 3(a), MD Rules: "academic clinical study" means a clinical study conducted for academic purpose on a medical device for the approved or a new intended use, new material of construction, new improved design or new population.
8 Rule 3 (l), MD Rules: "clinical investigation" means the systematic study of an investigational medical device in or on human participants to assess its safety, performance or effectiveness.
9 Rule3(n), MD Rules: "clinical performance evaluation" means the systematic performance study of a new in vitro diagnostic medical device on a specimen collected from human participants to assess its performance.
10 Rule 3(m), MD Rules: "predicate device" means a device, first time and first of its kind, approved for manufacture for sale or for import by the Central Licensing Authority and has the similar intended use, material of construction, and design characteristics as the device which is proposed for licence in India.
11 Rule 2 (1)(hh), NDCT Rules: "sponsor" includes a person, a company or an institution or an organization responsible for initiation and management of a clinical trial.
Rule 3 (zt), MD Rules: "sponsor" includes a person, an investigator, a company or an institution or an organization responsible for the initiation and management of a clinical investigation or clinical performance evaluation in India.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.

