
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- with readers working within the Healthcare industries
- within Wealth Management and Strategy topic(s)
What Are ADCs and Why Are They Growing?
Over the past few years economic headwinds have resulted in fewer deals, with companies and private equity firms alike reassessing where to spend money. But one portion of the pharmaceutical industry is bucking this trend in a major way. Since 2018, licensing deals totaling over US$60 billion have been signed regarding ADCs, with 2023 alone having at least 18 deals. As a result, worldwide antibody drug conjugates (ADCs) sales are projected to reach US$20-30 billion per year in the near future.
ADCs are a class of chemotherapy medicines used in the treatment of cancers. These drugs combine an antibody and cancer treating drug to minimize the impact of chemotherapy on the healthy tissue in patients, while maximizing their impact on the cancer. The antibody is chosen to target the specific cancer's cells and guide the cancer drug to the specific part of the body requiring treatment. Earlier cancer treatments were unable to localize the impact of the powerful chemotherapy drugs and caused more collateral damage in the body. ADCs do a better job of specifically targeting the part of the body being treated. This can also lead to a smaller dose of the cancer drug in the body, further reducing the overall negative impact on the patient while still treating the cancer.
ADCs are not a new technology, as the first ADC was approved in 2000 to treat leukemia. Continued development of the technology has given regulators, pharmaceutical companies, and doctors more confidence in the clinical efficacy of these treatments. More than 140 new ADCs are currently in clinical development.
The rapid expansion of this area of technology raises many business and legal questions. These topics include:
- Issues related to deals and licensing for ADCs/antibody therapeutics
- Patent/IP challenges to consider for ADCs/antibody therapeutics
- Clinical trial issues for ADCs/antibody therapeutics
- FDA/regulatory issues for ADCs/Antibody therapeutics
- Litigation issues for ADCs/antibody therapeutics
Antibody Drug Conjugates: An FDA Perspective
ADCs represent an innovative class of potent anti-cancer compounds. ADCs are widely used in the treatment of hematologic malignancies and solid tumors.
In contrast to conventional chemotherapeutic drugbased therapies that are mainly associated with modest specificity and therapeutic benefit, ADCs are composed of three critical components: (1) a monoclonal antibody; (2) bound to a cytotoxic drug; and (3) a chemical linker moiety. They are a class of targeted drugs composed of a payload linked to an mAbs (antibody) that is designed to specifically release their payload at a tumor site. ADCs are capable of achieving clinical benefit in terms of targeted killing of cancer cells and, while sparing healthy tissues, a reduction in systemic side effects caused by off-tumor toxicity. ADCs are being increasingly used in combination with other agents, including as first-line cancer therapies.
The antibody moiety targets a specific cell surface antigen expressed by tumor cells and/or cells of the tumor microenvironment. The antibody acts as a carrier that delivers the cytotoxic payload within the tumor mass. Notwithstanding, the development of ADCs has challenges, including: (a) low tumor selectivity when the target antigens are not exclusively expressed by cancer cells; (b) premature release of the cytotoxic drug into the bloodstream as a consequence of linker instability; and (c) development of tumor resistance mechanisms to the payload. These factors may contribute to a decrease in efficacy and/or in no safety improvement compared to unconjugated cytotoxic agents.
ADCs are subject to all pertinent laws and regulations for biological products, including those governing product development under Investigation New Drug exemptions (INDs), testing, and approval as outlined in section 351 of the Public Health Service Act (42 U.S.C. 262). In March 2024, the U.S. Food and Drug Administration (FDA) issued a guidance on the Clinical Pharmacology Considerations for Antibody-Drug Conjugates. FDA's Guidance specifically outlines clinical pharmacology considerations of ADC development programs.
The FDA Guidance includes regulatory development considerations information in the following areas:
- Key Considerations for ADC Dosing Strategies, including dosing strategies and external and intrinsic dosing considerations,
- Clinical Pharmacology, including bioanalytical approaches, dose and exposure response, intrinsic factors for consideration, and pharmacogenomics,
- Assessment of the product on the QTc interval, (a measurement made on an electrocardiogram used to assess some of the electrical properties of the heart),
- Immunogenicity, and
- Drug-Drug Interactions.
As is clearly evident from the comprehensive nature of FDA's guidance as well as the approvals included in the table below, ADCs offer a novel approach to targeted cancer therapy. The diversification of antigenic targets as well as bioactive payloads rapidly broadens the scope of tumor indications for ADCs. Moreover, novel vector protein formats as well as mAbs targeting the tumor microenvironment can be expected to improve the intratumor distribution or activation of ADCs, and consequently their anticancer activity. As with many oncology agents, toxicity remains a key issue in the development of ADCs and better understanding and management of ADC-related toxicities will be essential for further optimization.
These ADCs represent a significant breakthrough in the treatment of cancer. It is important that companies carefully consider their development programs in order to expedite the FDA review and approval process. Both companies and patients benefit from these new treatment modalities. Foley's team of FDA regulatory and clinical trial experts can help companies navigate through product development and FDA's submission, review and approval process.
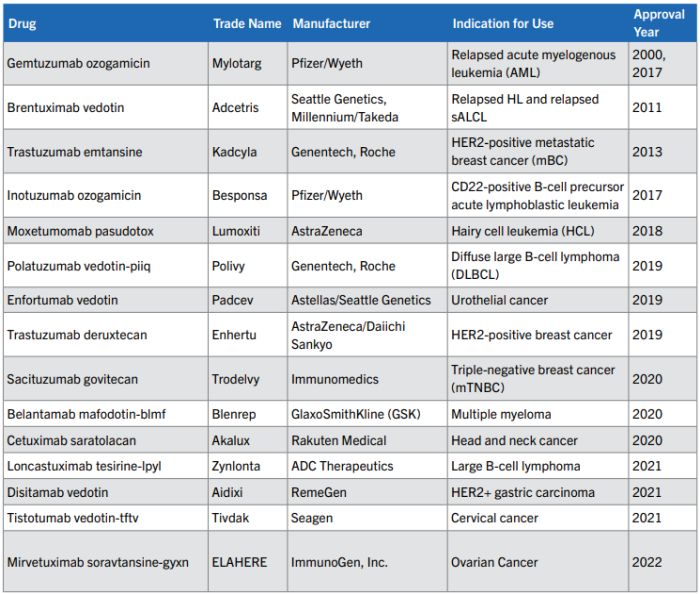
15 ADCs have been approved as of October 2023 for marketing by the FDA for use in clinical oncology as seen in the table below.
Strategies for Patenting Antibody-Drug Conjugate Inventions
ADC is a promising class of cancer treatments with accelerating FDA approval and rapidly growing market size as discussed in previous articles in this series. Despite the success of ADCs in treating cancer, securing patent protection for ADCs often presents unique challenges. This article discusses two strategic patent prosecution approaches to overcoming these challenges: identifying lack of motivation to combine the components of the ADC and demonstrating unexpected results.
I. Introduction
ADCs provide abundant opportunities for new cancer treatments, innovation, and collaboration across different industries because they combine three distinct technologies: (1) an antibody, (2) a toxic payload, and (3) a linker that joins them. While ADCs are based on the seemingly simple idea of using the targeting ability of antibodies to deliver more toxically potent drugs to specific cancer cells, ADCs are complicated molecules that pose significant technical, regulatory, and intellectual property challenges.
Indeed, development of an effective and safe ADC therapy requires significant research and innovation to ensure that the right components are combined in the right way to avoid side effects, inefficiency, tumor resistance, and pharmacokinetic profiles that make the drug delivery unpractical. In some cases, overcoming these challenges requires development of new generations of ADC modalities by using novel payloads, modified antibody backbones, and new drug linker-release mechanisms, as reviewed in Tsuchikama, K. et al., Nature Rev. Clin. Oncol., 21, 203– 223; 2024.
Even though the development of an effective ADC requires intense research to find the right combination, Patent Offices or Courts may find ADCs to be an "obvious" combination in cases where the components of the ADC were previously used for the same purpose as the ADC. Obviousness may present a major obstacle for patent protection of ADC innovations if the patent claims are not sufficiently supported by evidence and patent prosecution strategies.
II. Strategies for Addressing Obviousness Challenges to ADC
The key strategies of overcoming obviousness challenges to ADC inventions (section II a)) are demonstration of lack of motivation to combine the parts of the ADC (section II b), and evidence of unexpected results achieved by the ADC (section II c)).
a. The Legal Standard for Obviousness
The current framework for analyzing obviousness was established by the Supreme Court decision in KSR International Co. v. Teleflex Inc., 550 U.S. 398, 418 (2007). According to KSR, when a claimed invention is rejected for obviousness because the invention appears to be a combination of known elements, the Examiner must, "identify a reason that would have prompted a person of ordinary skill in the relevant field to combine the elements in the way the claimed new invention does." See KSR, 550 U.S. at 401. The Federal Circuit further explained in post-KSR decisions that the mere plausibility of prior art combinations is insufficient to establish a prima facie case of obviousness. See PersonalWeb Tech v. Apple, 848 F.3d 987, 994 (Fed. Cir. 2017) stating that:
[The] reasoning seems to say no more than that a skilled artisan, once presented with the two references, would have understood that they could be combined. And that is not enough: it does not imply a motivation to pick out those two references and combine them to arrive at the claimed invention.
A prima facie case of obviousness may be rebutted by submitting objective evidence that the claimed invention provided unexpected results. The post-KSR decision in Bristol-Myers Squibb Co. v. Teva Pharmaceuticals USA, Inc., 752 F.3d 967, 977 (Fed. Cir. 2014) determined that "particularly probative" evidence of unexpected results "establish[es] that there is a difference between the results obtained and those of the closest prior art, and that the difference would not have been expected by one of ordinary skill in the art at the time of the invention."
Thus, an ADC should not be held obvious merely because the different components of the ADC were previously known. Rather, the ADC can be held obvious if sufficient guidance or motivation to combine the specific components the same way as in the claimed ADC has been established. Even if there is motivation to combine known components into a specifically claimed ADC, the ADC may still be patentable if the ADC provides unexpected results. Therefore, patenting ADCs with known components will often hinge on finding reasons a skilled artisan would not combine the specific ADC components or showing that the particular ADC yielded unexpected results compared to the components, as further discussed and illustrated below.
b. Demonstrating a Lack of Motivation to Make the Claimed Combination
1. ADC therapy claims may be nonobvious because they recite features that the antibody portion of the ADC failed to perform by itself.
Patent claims covering a particular ADC therapy may be found nonobvious due to a lack of motivation to combine known elements into the claimed ADC if the antibody and/ or conjugate failed to treat a particular patient population individually. For example, U.S. Patent 7,575,748 survived an Inter Partes Review (IPR) challenge based on obviousness because the claimed ADC could be used to treat a particular indication that the antibody portion of the ADC could not treat by itself as highlighted in the quoted claim language below. See Phigenix, Inc. v. Genentech, Inc. and Immunogen, Inc., IPR2014-00842. The allowed claims in U.S. 7,575,748 recited:
A method for the treatment of a tumor in a mammal, comprising the steps of
(i) identifying said tumor as being characterized by overexpression of an ErbB2 receptor and as being a tumor that does not respond, or responds poorly, to treatment with an anti-ErbB antibody, and
(ii) intravenously administering to the mammal a therapeutically effective amount of a conjugate of a humanized antibody huMab 4D5-8 covalently linked via a thioether linking group with a maytansinoid DM1....
U.S. Patent 7,575,748 illustrated that method claims covering an ADC formed by known components can be found allowable based on properties of the ADC.
2. Establishing that a component of the ADC was discouraged
U.S. Patent 8,337,856 is directed to ADC composition claims, and upon challenge the claims were held valid even though they recited known antibodies and conjugates. See Phigenix, Inc. v. Immunogen, Inc., Case IPR-2014-00676, Final Written Decisions dated October 27, 2015 (Paper 39). Independent claim 1 of the '856 patent recites:
1. An immunoconjugate comprising an anti-ErbB2 antibody conjugated to a maytansinoid, wherein the antibody is huMAb4D5-8.
huMAB4D5-8 was commercialized in the prior art product, Herceptin®, and used for treating breast cancer in combination with other cytotoxic agents. In addition, maytansinoid had already been used as a conjugate to different antibodies. The patent challenger, therefore, argued that it would be obvious to use Herceptin® with maytansinoid.
However, the patent owner successfully presented evidence suggesting to a skilled artisan that "Herceptinmaytansinoid immunoconjugates would have been expected to exhibit unacceptable levels of antigendependent toxicity in normal human liver tissue in patients." See pages 16–22 of IPR-2014-00676 (Paper 39). The Board found this argument persuasive because the patent challenger had not explained that an ordinary artisan would have been motivated to make the claimed ADC, given the reported liver toxicities of maytansinoid immunoconjugates.
Given these cases, strategies for overcoming obviousness rejections could be found by determining if:
- There are known problems of toxicity associated with the toxic payload component of the ADC or other reasons not to use the toxic payload as claimed, and
- The antibody portion of the ADC has previously been reported to be ineffective by itself against the claimed indication.
As will be discussed further below, the claims of U.S. Patent 8,337,856 were also found valid because of unexpected results, which is another central strategy for obtaining patent coverage of ADCs.
c. Unexpected Results Achieved by the ADC
1. Example of ADC claims found valid based on unexpected results
The claims of U.S. Patent 8,337,856 were found valid based on unexpected results even though the components of the claimed ADC were known. See Immunogen, Inc., IPR-2014-00676 (Paper 39). In particular, the Board held that the Herceptin-maytansinoid immunoconjugate claims were non-obvious because the patent owner provided substantial evidence of unexpectedly superior results compared to the "closest prior art" composition. See pages 23-25, Immunogen, Inc., IPR-2014-00676 (Paper 39). The "closest prior art" was the antibody by itself, and the results demonstrated that the ADC overcame some limitations of the "naked" antibody.
2. Example of evidence of unexpected results not sufficient to establish nonobviousness
Demonstrating unexpected results can be challenging, as shown in Hospira v. Genentech, IPR2017-00731 (Paper 120, at page 23 (PTAB October 3, 2018)), where the claims of U.S. Patent No. 7,846,441 were found unpatentable. The claims of U.S. Patent No. 7,846,441 are represented by claim 1:
1. A method for the treatment of a human patient with a malignant progressing tumor or cancer characterized by overexpression of ErbB2 receptor, comprising administering a combination of an intact antibody which binds to epitope 4D5 within the ErbB2 extracellular domain sequence and a taxoid, in the absence of an anthracycline derivative, to the human patient in an amount effective to extend the time to disease progression in said human patient, without increase in overall severe adverse events.
The ADC covered by this patent contains an antibody that binds to epitope 4D5 within the ErbB2 extracellular domain sequence and a taxoid. A previous publication asserted against this patent disclosed the same antibody conjugated with a taxoid tested in a mouse model. The patent owner argued that the claimed method yielded unexpected results in humans as compared to the previous mouse studies. However, the Board held that a mouse study is a "reliable predictor of success in humans," and the results from the mouse study in prior art predict that the ADC would also be effective in humans. See Hospira v. Genentech, IPR2017-00731 (Paper 120, at page 26 (P.T.A.B. Oct. 3, 2018).
In addition, the Board in Hospira v. Genentech found the patent owner's statements to the FDA to be evidence of obviousness of the claims. The patent owner had requested FDA approval of a combination of an antibody binding ErbB2 (trastuzumab) and a taxoid (paclitaxel) because a skilled artisan would expect that this combination was effective based on the same prior art as asserted against U.S. Patent 7,846,441. Hence, the Board considered the patent owner's statements at the FDA as evidence that the results obtained with the claimed ADC were expected. See Hospira v. Genentech, IPR2017-00731 (Paper 120, at pages 27-28 (P.T.A.B. Oct. 3, 2018).
The argument for nonobviousness based on unexpected results in Hospira v. Genentech failed because the closest prior art was a combination of the same components as the claimed ADC. Accordingly, in cases where the closest prior art to a claimed ADC only discloses one of the components of the ADC, the availability of beneficial results obtained with the ADC will help obtaining allowance of the claims or surviving a challenge based on obviousness.
Main Takeaways
Based on the above review, we provide the following considerations for determining strategies for obtaining patent coverage of ADCs in cases where the components of the ADC are known:
- Is use of one of the ADC components discouraged for the claimed method (e.g., would it be too harmful)?
- Could the selected payload interfere with the properties of the antibody or vice versa (e.g., cause aggregation, lower solubility, or changes to important post-translational modifications of the antibody)?
- Have the components of the ADC been used for treating the same indications or is there a lack of guidance for using one or more of the ADC components as claimed (i.e., lack of motivation)?
- Is objective evidence of unexpected results from the ADC available (such as improved efficacy or tolerability as compared to individual components)?
Finally, it should be noted that the linker chemistry is also an important part of the ADC and patentability of the ADC. The ADC linker was not central to the above discussed cases because it was not recited by the independent claims. However, the Board did analyze dependent claims reciting specific non-cleavable linkers IPR-2014-00676, which would further distinguish U.S. Patent 8,337,856 from prior art. Accordingly, linker chemistry may confer patentability to an ADC.
Indeed, the linker chemistry offers fertile grounds for innovation and development of new generations of ADCs. See Tsuchikama, K. et al., Nature Rev. Clin. Oncol., 21, 203–223; 2024. The linker chemistry can, for example, be used to regulate the amount of toxic payload (i.e., the drug-antibody ratio), and when and where the payload is released to improve the efficacy and tolerability of the ADC. Even though the linker chemistry by itself may not be new, there may be a lack of motivation to use a particular linker strategy in an ADC because the linker interferes with the antibody structure, changes post-translational modification of the antibody, causes aggregation, or provides too high or too low payload-antibody ratio. Thus, a careful consideration of the linker and its effects on the function of the ADC could also be critical for obtaining patent protection of the ADC.
Ultimately, successful strategies of patenting ADCs will depend on the results obtained with the ADC, and what results would be expected based on common knowledge of the antibodies, toxic payloads, and linker chemistry that make up the ADC.
Deals and Licensing for Antibody-Drug Conjugates
ADCs are a promising class of cancer treatments with an accelerating number of U.S. Food and Drug Administration (FDA) approvals and rapidly growing market size, as discussed in previous articles in this series. This article discusses issues relating to deals and licensing for ADCs/ Antibody therapeutics.
Introduction
About 10 million people die from cancer every year, making it one of the largest health problems globally. Chemotherapy has remained a hallmark of cancer treatment since the 1940s. However, traditional chemotherapy is a "blunt force instrument" that not only kills fast-growing cancer cells, but also kills healthy cells that grow and divide quickly, such as mucosal cells which line the mouth and gastrointestinal tract, as well as hair cells. Some chemotherapy toxicity, such as fatigue, neuropathy, hair loss and heart damage, can last a lifetime.
ADCs, sometimes called "smart chemotherapy", are cancer therapeutics comprising three blocks: a selective monoclonal antibody, a stable linker, and a potent cytotoxic drug. With an ADC, the antibody identifies biomarkers on cancer cells and attaches itself to them, the chemical linker then breaks, enabling delivery of the cytotoxic drug payload into cancer cells without collateral damage to healthy cells. Compared to conventional cancer therapies, ADCs improve treatment outcomes with respect to tumor remission, time to tumor progression, and overall survival by specifically channeling cytotoxic agents into the malignant target cells, thereby limiting the exposure of healthy tissue to adverse effects. Thus, ADCs can dramatically reduce chemotherapy toxicity as well as open the door to the use of new highly toxic chemotherapeutic agents.
The efficacy of the 14 ADCs currently approved in the U.S. (Mylotarg", Adcetris", Kadcyla", Besponsa", Lumoxiti", Polivy", Padcev", Enhertu", Trodelvy", Akalux", Zynlota", Aidixi", Tivdak", Elahere") is derived from the introduction of novel linkers for the binding of cytotoxic agents to the antibody as well as the development of new, potent cytotoxic agents. Such potent cytotoxic agents, in the absence of antibody targeting to cancer cells, can exhibit a toxicity level which prohibits conventional use.
Oncology is the largest and the fastest-growing therapeutic area for biopharma, and this has triggered an increased flow of M&A investment towards ADCs. In 2023, ADCs were valued at US$9.7B in market revenue, and ADC revenue is expected to grow to US$19.8B by 2028. At present, there are over 60 biopharma companies involved in the ADC space and at least 100 ADC drugs in clinical trials.
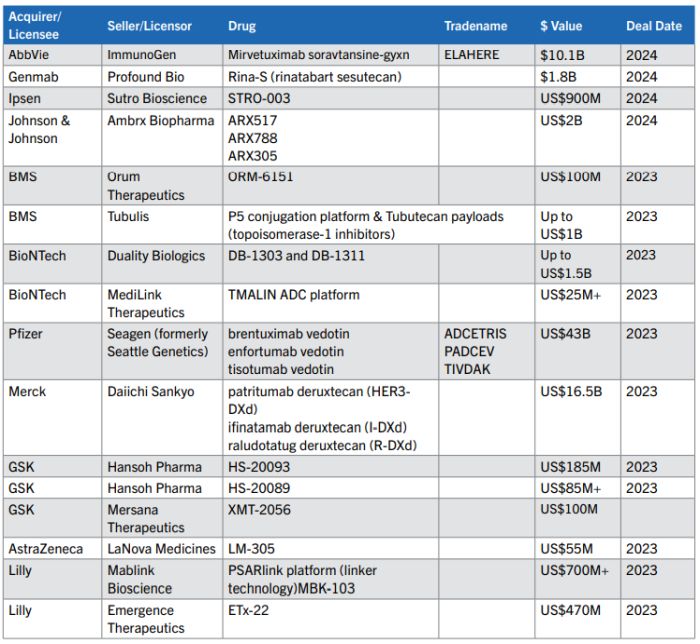
This rapid pace of innovation has spurred dealmaking in the ADC tech space. In 2023, there were 76 ADC deals made — including licenses, collaborations and acquisitions, and the deal pace is continuing in 2024 (see the table below with exemplary deals listed).
Issues that Impact Deal Valuation
1. Nature of the technology to be licensed
Is the technology to be licensed platform technology applicable to a number of ADC drugs, such as a new linker technology, or it is specific to one or more drug candidates? Platform linker technology has significant value, but it does not have the same value as a marketed drug.
Although the concept seems simple, the combination of three components of ADCs (monoclonal antibody, linker, and cytotoxin) into an optimized and functional therapeutic agent remains a great challenge:
- Linker: If the technology relates to an ADC linker, is there extensive data demonstrating the improved efficacy of multiple ADCs utilizing the linker?
- Payload: The first generation of ADCs used classical chemotherapy drugs such as doxorubicin and methotrexate with the benefit of a well-known toxicity profile. Newer ADCs utilize potent tubulin inhibitors, DNA damaging agents, and immunomodulators, but these drugs tend to have a less robust toxicity profile. Notwithstanding the targeted delivery of ADCs, only about 2% of ADCs reach targeted tumor sites after intravenous administration, thus making payload toxicity a concern.
- Antibody targeting antigen: What is the identity of the target cancer antigen, typically specific proteins overexpressed in cancer cells, such as HER2, trop2, nectin4 and EGFR in solid tumors, and CD19, CD22, CD33, CD30, BCMA and CD79b? Newer ADCs utilize bi-specific antibodies targeting multiple antigens to improve specificity and immune response.
2. Stage of development
If the technology relates to an ADC drug candidate, at what stage in development is the ADC? Early-stage development ADCs carry an inherently higher risk, and therefore lower valuation, although even late-stage development ADCs can fail. For example, AbbVie first entered the ADC space in 2016 with the acquisition of Stemcentrx, but subsequently the ADC Rova-T (Rovalpituzumab Tesirine) failed clinical trials. Similarly, following GSK's 2023 acquisition of Mersana Therapeutics' XMT-2056, a clinical hold was put on the drug following a patient death, although the hold was lifted by the FDA about 7 months later in late 2023. Further, two ADCs approved by the FDA, Mylotarg" and Blenrep", had their approvals withdrawn due to failure to meet requisite endpoints in post-approval trials. Mylotarg" was subsequently re-approved at a lower dose in combination with chemotherapy.
3. Competitive products, disease indication, potential patient population
For the technology encompassed by a potential license or acquisition, are there competitive ADC products on the market or in late-stage clinical development, thereby shrinking the potential patient population? Also, there is a higher threshold for U.S. FDA approval for an ADC having the same indication as a prior approved ADC. Notably, none of the current FDA approved ADCs have the exact same indication.
Is the potential disease indication particularly challenging to treat? Abbvie's failed Rova-T ADC was being evaluated for small cell lung cancer, which is a particularly recalcitrant type of cancer. To be clear, pharma companies should not shy away from seeking treatments for challenging types of cancer, but deal valuation should recognize the difficulty of success with treating certain cancer types. Another factor in the Rova-T studies was that the Rova-T development strategy moved directly from promising small phase 1 studies to large phase 3 studies without confirming the safety and efficacy data in phase 2 studies. It's understandable that patients are eager for new treatments, but taking shortcuts in development can have undesirable consequences, and these factors should impact deal valuation.
What is the potential patient population? Certain cancers qualify for orphan drug designation, meaning a rare disease or condition impacting less than 200K patients in the United States. In 2024, the FDA granted orphan drug designation to Mabwell's ADC 9MW2821 for the treatment of patients with esophageal cancer; in 2023 the FDA granted two orphan drug designations to Antengene's ADC ATG-022 for the treatment of patients with gastric cancer and pancreatic cancer; and in 2022 the FDA granted orphan drug designation to Mersana Therapeutics' ADC XMT-2056 for the treatment of gastric cancer. Orphan drug designation provides multiple benefits, such as exemption from FDA user fees, tax incentives for clinical trials, smaller clinical trials, minimal competition, and potential for seven years of market exclusivity after approval.
Therapies for cancers which occur more frequently than orphan drugs are also highly desirable. For example, breast cancer is the most common cancer type with 313,510 new cases expected in the United States in 2024, and there are multiple ADCs approved for treating different types of breast cancer: Kadcyla" (HER2-positive metastatic breast cancer (mBC)), Enhertu (HER2-positive breast cancer), and Trodelvy (triple-negative breast cancer (mTNBC)). Notwithstanding these treatments, and that breast cancer deaths are decreasing, breast cancer is still the second leading cause of cancer death in women so there remains a need for new and improved therapies.
4. Exclusivity & Freedom to Operate
Is the technology proprietary and protected by a robust patent portfolio? While the pace of development for ADCs is rapidly accelerating, the timeline from initial research and development to market launch is not short and the capital investment needed to conduct initial research and clinical trials is extensive. Thus, it is critical that ADCs in development be protected by a robust patent portfolio. A more extensive discussion regarding patenting of ADCs can be seen in our recent "Cancer Drugs: Strategies For Patenting Antibody-Drug Conjugate Inventions" article.
A flip side to exclusivity is whether there is freedom to operate (FTO) for the licensed technology. An FTO assessment for an ADC needs to address the multiple components of the licensed or acquired ADC.
5. Commercial relationship between the parties
The presence of an existing commercial relationship between two parties can positively impact valuation of a new deal. See the multiple deals between GSK and Hansoh Pharma, with the first deal having a valuation of US$86M and the second a valuation of US$185M. An existing relationship reduces risk in that the parties are a known quantity to each other, and subsequent deals build upon technology and research following an initial agreement.
6. Market pricing
Finally, a factor that supports increased valuation of ADC deals is the existence of multiple FDA approved ADC products on the market, meaning that pricing of ADCs is not new. It is always hard to be the first to launch a new type of therapeutic (see e.g., the pricing controversy regarding Solvaldi" for treatment of Hepatitis C). Pricing of ADCs needs to factor in the complex manufacturing, as well as R&D for multiple failed ADCs. Current U.S. list pricing of ADCs include:
- US$19,231 per cycle (2 infusions) for Gilead's Trodelvy"
- US$18,500 -US$25,000 per month for ImmunoGen's Elahere"
- US$34,000 per month for Pfizer's Tivdak" (21-day cycle)
- US$27,703 for one intravenous injection lpyl 10 mg of ADC Therapeutics' Zilina"
- US$13,300 per month for AstraZeneca's Enhertu" (21-day cycle)
- US$17,718 per patient per 28-day cycle for Astellas' Padcev"
- US$22,450 per month for Roche's Polivy" (21-day cycle)
Antibody-Drug Conjugate Litigation
ADCs are typically composed of a monoclonal antibody attached to a cytotoxic drug via a chemical linker. The antibody is able to identify biomarkers on and attach to cancer cells, allowing targeted delivery of the cytotoxic drug without damaging surrounding, healthy cells. As a result of this targeted treatment, ADCs are associated with improvement in tumor remissions and overall survival, as well as decreased side effects while undergoing treatment.
Although the first ADCs came to market two decades ago, the technology proved difficult to translate into clinical administration. For this reason, while ADCs existed, there were not many products in the market, and thus little litigation surrounding the technology. In recent years, there has been a resurgence in ADC development due to improvements in cytotoxic agents, linker technologies, and the potential to combine ADC treatment with immunotherapy and chemotherapy. There are currently 16 ADC drugs that are FDA-approved and have launched, but the pipeline is robust—there are over 250 ADCs in preclinical development, and nearly another 150 in some stage of clinical development. As the market continues to mature, with more companies and products securing global approvals, there will likely be a concurrent increase in litigation involving ADCs.
Patent Litigation and Other Intellectual Property (IP) Issues
The area where there has been the most litigation activity has been surrounding patents for ADCs. This makes sense, as the first step to commercializing a product and making it available to the clinical population is often obtaining patent protection. A previous article in this series highlighted potential strategies for patenting ADC inventions. With increasing numbers of ADC patents granted, it is logical to expect that litigation alleging infringement of these patents will continue to rise.
These litigations could result from patent infringement when one company's claims are infringed by a competitor working on a similar product. Given the current focus on certain diseases, such as cancer, we will likely see numerous companies targeting similar or identical antigens on cells. Consequently, two companies may have claims covering similar types of antibodies in the ADC complex and this could lead to litigation.
Similarly, companies may develop new types of linker systems for their ADCs which are incorporated into competitor products. When this occurs, there may be patent claims from the original developer which are infringed by competitors using a similar linker system.
Given the enormous potential of ADCs, companies may also partner together in the research and development and clinical trials phases to develop technology. Some of these partnerships will terminate, but each company will continue development of new products as well as pursuit of IP covering their work. Later, one company may sue the other for infringing patent claims and the companies will dispute whether the underlying IP was jointly developed during the partnership or afterwards. It's critical for partnerships to clearly agree on ownership of jointly developed IP in order to avoid these scenarios.
Finally, an increase in patent litigation will also lead to increased IPR (Inter Partes Review) filings at the Patent Trial and Appeal Board (PTAB). Any time a patent infringement suit is filed, defendants consider whether or not to challenge the patent at the United States Patent and Trademark Office (USPTO) using an IPR. With the increase in ADC litigations, we will likely see more focus on ADC patents at the PTAB as well.
Other Potential Litigation Issues
Due to the relatively untested waters in the ADC arena, there has not been extensive litigation involving these molecules. However, as the market continues to grow and more patients begin to use ADCs, there will likely be lawsuits involving ADCs in much the same way as any other pharmaceutical product.
For example, drug products are easily susceptible to product liability actions. Pharmaceuticals typically have side effects, and the same is true of ADCs. Where patients experience unexpected side effects or adverse events when using a drug, the manufacturer may be at risk of a lawsuit for negligence, failure to warn, misleading labeling, defective manufacturing, or improper marketing.
ADCs could also become more prominent players in lawsuits regarding Medicare negotiation of pricing for drugs. Under the Inflation Reduction Act (IRA), Medicare has the authority to negotiate prices directly with pharmaceutical and biotechnology companies, but only with respect to certain drugs that do not have generic or biosimilar equivalents and that have been on the market for a set number of years. Drug companies like Merck have filed lawsuits challenging this provision of the IRA as unconstitutional and impermissibly stifling innovation.
ADCs are likely to fall into this category as the market matures, as they are unlikely to have biosimilar equivalents—that is, a product with no relevant differences from an already-approved biotherapeutic (known as a reference product) regarding efficacy, safety, quality, purity, and potency. Because of the vastly complex nature of the monoclonal antibody component of an ADC, experts agree that it is unlikely that any ADC will receive approval through the biosimilar process in the near future. Therefore, ADCs on the market may be subject to price negotiation with Medicare, leading to further future lawsuits over the IRA as it applies to ADCs.
Clinical Trial Issues for Antibody Drug Conjugates (ADCs)/ Antibody Therapeutics
ADCs are a class of small molecule drugs (also known as a payload) and an antibody conjugated together by a chemical linker. ADCs are designed to target specific cells, such as cancer cells, while minimizing the impact on nontargeted, healthy cells. The goal of ADCs is to maximize efficacy and minimize systemic toxicity.
The FDA has already approved several ADCs and more are currently under development, with clinical trials being conducted in relation to same. As discussed in our prior article on the FDA Perspective of ADCs, ADCs are subject to relevant laws and regulations for biological products, including those governing product development under Investigation New Drug exemptions (INDs), testing, and FDA review and approval prior to commercialization and marketing. The FDA approval process includes and necessitates the conduct of clinical trials, which trials need to be designed and conduced so as to be capable of producing clinical data supportive of FDA approval.
In May 2024, FDA issued guidance on the Clinical Pharmacology Considerations for Antibody-Drug Conjugates to address a range of considerations necessary for ADCs under development. While ADCs are subject to the typical laws and regulations for biological products, in the issued guidance FDA emphasized that ADCs are distinct from both biologics and small molecule drugs, requiring special considerations throughout the development process due to their unique structure and mechanism of action.
Accordingly, careful consideration should be given to the design and conduct of clinical trials involving ADCs due to the importance of generating comprehensive clinical data necessary to obtain the investigational drug's approval.
Drug Development Timeline
FDA's recommendations in the guidance are applicable during the development phase, commencing with preclinical development through clinical development and Phase 1, 2, and 3 trials.
In its guidance, FDA indicates that information and data collected in preclinical assessment and early studies will inform the ADC's development strategy and the overall design of later stage studies. For example, FDA provides that an in vitro Drug-Drug Interactions (DDI) risk assessment should inform the need for, and design of, in vivo DDI studies. The absorption, distribution, metabolism, and excretion information from the preclinical and early clinical studies will inform whether intrinsic factors, such as organ impairment, should be evaluated in pivotal studies or in dedicated studies.
ADC Development Considerations
The guidance covers a wide range of considerations when designing ADC studies including:
1. Dosing Strategies
2. Bioanalytical Approach
3. Dose- and Exposure-Response
4. Intrinsic Factors
5. QTc Assessment
6. Immunogenicity
7. Drug-Drug Interactions
Many of these considerations impact early stages of development and FDA may recommend conducting additional risk assessments or validations that exceed what is typically required for other drugs early on in the drug development process to ensure a thorough evaluation of the investigational drug and its constituent parts. For example, FDA recommends conducting a multi-tiered immunogenicity assessment to evaluate immunogenicity to ADCs and the potential impact on pharmacokinetics (PK), safety, and efficacy because ADCs tend to have a relatively narrow therapeutic range. FDA may also require multiple assays to evaluate where the anti-drug antibodies bind to.
Clinical Trial Considerations
Because of the increased requirements that commence and are applicable to the early stages of development, it is critical that developers of ADCs carefully structure their drug development programs to ensure that all preclinical studies, along with Phase 1, 2, and 3, studies are appropriately capturing the level of detail and thoroughness which FDA will require when reviewing an IND, and ultimately, an NDA. It is imperative that developers of ADC maintain a strong working relationship with FDA throughout this process and stay informed regarding any new guidance that may impact ADC development.
In addition, ADC developers should establish strong compliance programs, such as conducting frequent internal auditing and maintaining robust standard operating procedures, beginning as early as prototype design and discovery to ensure that the developer, as a clinical trial sponsor, meets all quality system and other FDA requirements.
ADC developers should also pay careful attention when negotiating clinical trial agreements to ensure all early stage preclinical and clinical data, information, and results are protected, confidential, and owned by the sponsor.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]






