
- within Food, Drugs, Healthcare, Life Sciences and Corporate/Commercial Law topic(s)
- in United States
Research assistant Nicole Gebert contributed to this article.
After having discussed the general purpose and background of the IVDR, the new definition of IVDs, and the conformity assessment in light of the new risk classification as well as the role of notified bodies in part 1 of our 3 part series on the entering into force of the EU In Vitro Diagnostic Regulation (EU) 2017/746 (IVDR), we will now explore key changes in the clinical evidence, post-market surveillance.
CLINICAL EVIDENCE, PERFORMANCE EVALUATION, AND PERFORMANCE STUDIES
Ensuring that the clinical evidence can stand up to the scrutiny of the notified bodies will be one of the biggest challenges for manufacturers in IVDR compliance. It is the responsibility of the manufacturers to determine whether they have adequate clinical and performance data to demonstrate conformity with the relevant safety and performance requirements. According to the IVDR, clinical evidence must demonstrate a continuous process of performance evaluation and must support the intended purpose of the device as stated by the manufacturer. Gradually, the level of clinical evidence needed to demonstrate device conformity increases in line with the risk class. A manufacturer must document the clinical evidence in a performance evaluation report once they are satisfied they have all of the data needed to support compliance with the IVDR. The report must include the scientific validity report, the analytical performance report, the clinical performance report, and an assessment of those reports allowing demonstration of the clinical evidence. More detailed information on clinical evidence can be found in the MDCG Guidance on general principles of clinical evidence for In Vitro Diagnostic medical devices (IVDs) from January 2022.
POST-MARKET SURVEILLANCE
Similar to the MDR, the IVDR too places a greater emphasis on life-cycle management and continuous product evaluation than the IVDD. In this context, manufacturers are required to have a quality management system (QMS) in place (Article 10 IVDR). As part of the QMS, manufacturers are obligated to plan, establish, document, implement, maintain, and update a post-market surveillance system for each device in a manner that is proportionate to the risk class and appropriate for the type of device (Article 78 (1) IVDR). The requirements for the post-market surveillance system are described in Articles 79-81, Annex III, and Annex XIII of the IVDR. The requirements vary according to the risk class. Many aspects of the IVDR post-market surveillance requirements resemble the requirements of the MDR (for more information on QMS and post-market surveillance under the MDR, please see our previous blog post here). Similar to the MDR's post- market clinical follow-up, the IVDR stipulates that a post-market performance follow-up (PMPF) is required to ensure that an IVD's safety, performance, and clinical evidence remain based on factual evidence throughout its expected lifespan and that its benefit-risk ratio remains acceptable. In some cases, PMPF can be justified as not required (see Annex III 1b and Annex XIII Part B 8).
UNIQUE DEVICE IDENTIFICATION SYSTEM
The identification and traceability of IVDs will be improved under the IVDR with the introduction of unique device identifiers (UDI). As laid out in Article 24 IVDR, each IVD must have a UDI composed of two parts: a UDI that is specific to a manufacturer and a device that provides access to the information set forth in Part B of Annex VI IVDR, and a unique device production identifier that identifies the unit of device production and, if applicable, the packaged devices, as specified in Part C of Annex VI IVDR. Manufacturers are responsible for entering and maintaining the necessary data in a UDI database, which is part of the European Database on Medical Devices (EUDAMED). The UDI requirements in the IVDR are in line with the requirements set out by the MDR, which we addressed in more detail in part 5 of our MDR blog series that can be found here.
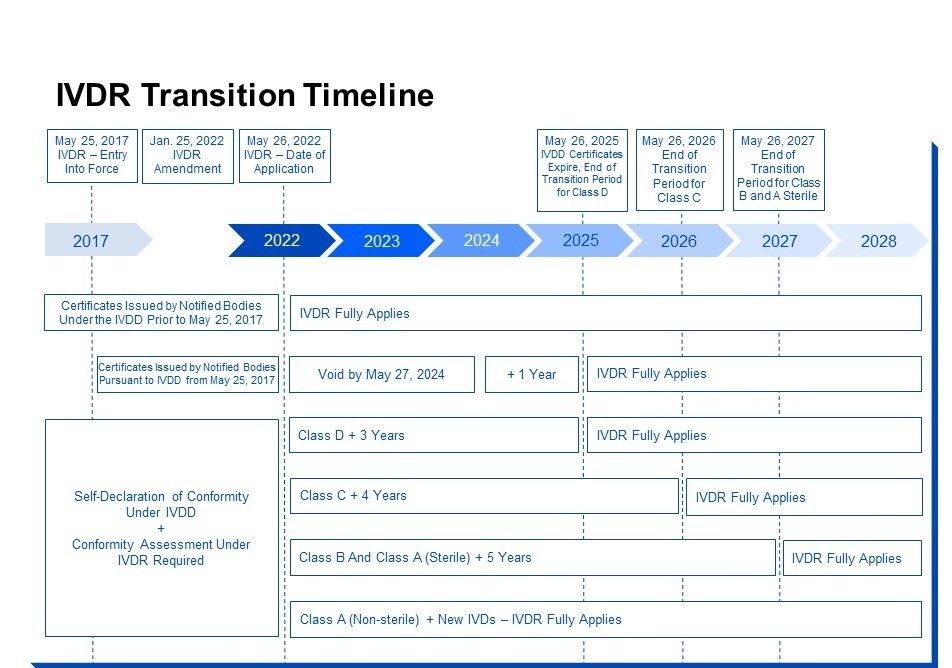
TRANSITION PERIODS
As mentioned above, the Commission proposed a delayed, progressive roll-out of the IVDR in October 2021. The rationale behind this proposal is that due to the resources diverted to address the COVID-19 pandemic and the significant changes introduced by the IVDR, Member State authorities, healthcare facilities, notified bodies, and economic operators would not be able to fully comply with the requirements of the IVDR at the time of application on May 26, 2022. In particular, the inadequate number of notified bodies was highlighted, making it impossible to guarantee that the conformity assessments would be carried out in time. After immense stakeholder pressure, the Commission acknowledged that this situation could lead to a significant disruption in the supply of a multitude of IVDs on the market both for health institutions and for the public (Commission proposal, press release, and Questions & Answers dated October 14, 2021). The proposal was adopted by the European Parliament in December 2021 (Commission press release, EU Parliament Resolution of December 15, 2021). Under the amended regulations, the dates on which certain requirements might apply are revised.
The extended transition periods apply only to legacy IVDs, meaning devices that were certified or declared compliant before May 26, 2022. Therefore, all devices newly placed on the market after May 26, 2022, must be classified and certified in accordance with the IVDR rules.
Furthermore, the IVDR will also immediately apply to any device for which an IVDD declaration of conformity was drawn up before May 26, 2022, and which must be reclassified as a class A non-sterile device under the IVDR. This is because such a device does not require a notified body review and, according to the rationale behind the transition periods, will thus not contribute to the notified bodies becoming a bottle neck that slows down market access or being subject to the same.
If a manufacturer has drawn up an IVDD declaration of conformity without the involvement of a notified body before May 26, 2022, and if, under the IVDR reclassification, the device must be classified as a class A sterile device or higher and thus undergo conformity assessment with a notified body, according to the amended Article 110 (3) IVDR, the following new transition periods apply:
- May 26, 2025, for class D devices;
- May 26, 2026, for class C devices;
- May 26, 2027, for class B devices; and
- May 26, 2027, for class A devices placed on the market in sterile condition.
For IVDs which have been certified by an IVDD notified body before May 26, 2022, according to Article 110 (2), the following transition periods apply:
- Certificates issued by notified bodies under the IVDD prior to May 25, 2017, will remain valid until the date indicated on the certificate (with no amendment of the transition period). It should be noted, however, that IVDD certificates are usually valid for a maximum of five years according to Article 9 (10) IVDD.
- Certificates issued by notified bodies in accordance with the IVDD from May 25, 2017, and certificates issued under the EC Verification in accordance with Annex VI of the IVDD, will become void at the latest by May 27, 2025.
It should be noted that for the aforementioned transition periods to apply, the IVD needs to continue to comply with the IVDD requirements and must not undergo significant changes in its design or intended purpose after May 26, 2022, which in particular for IVD software can be challenging because changes in the intended user groups or changed indications, corrections of errors to minimize risks, changes in the user interface (like changed warning messages), or the use of a different runtime environment will likely be deemed significant according to MDCG 2020-3 Guidance on Significant Changes. Furthermore, according to Art. 110 (3) IVDR, the IVD manufacturer must comply with post-market surveillance, vigilance, UDI, and market surveillance rules of the IVDR.
The transition periods can be summarized as follows:
CONCLUDING REMARKS
The IVDR brings about increased standards and requirements for IVDs in order for the devices to be placed on the market. Although it can be expected that many legacy IVDs, including IVD software, will benefit from the transition periods, manufacturers will need to assess on a case-by-case basis whether the respective device fulfills the criteria and whether the same can be maintained during the entire transition period (i.e., no significant changes can be made). Furthermore, the transition periods may bring some relief with respect to the certification of IVDs, but they do not necessarily mean that the IVDR requirements do not apply to these devices. Therefore, manufacturers should have adopted IVDR compliance measures by now and should seek. As it can be expected that the run on notified bodies will increase with the end of the transition periods approaching, it would also be advisable to start preparing the transition to IVDR certification and file for the same well in advance of the end of the applicable transition period.
Because of the generality of this update, the information provided herein may not be applicable in all situations and should not be acted upon without specific legal advice based on particular situations.
© Morrison & Foerster LLP. All rights reserved

