

- in United States
- within Tax, Intellectual Property and Law Practice Management topic(s)
A hostile authoritarian government launches a coordinated ransomware attack on computers across the globe.
In less than 24 hours, more than 200,000 computers in over 150 countries are infected, shutting down everything from MRI scanners to blood-storage refrigerators. Patient-filled ambulances are diverted and doctors at the operating table are left in limbo.
The narrative sounds like a New York Times best-selling political thriller.
But it describes the real-life 2017 WannaCry ransomware attack that caused thousands of lives to be put at risk and an estimated $4 billion in economic damage.
The WannaCry attack was instigated by a cyber hacking group that exploited vulnerabilities in outdated Microsoft Office systems to encrypt critical information as ransom for payments of Bitcoin. The U.S. believes the attack was instigated on behalf of the North Korean government presumably in retaliation for economic sanctions related to its nuclear program.
Unsurprisingly, medical device unavailability due to ransomware threats is now one of the FDA's top cybersecurity risk priorities. The risk is great and the threats are growing.
An estimated 20-30 billion medical devices in the U.S.—from infusion and insulin pumps to pacemakers and ventilators—are now connected to the Internet of Things (IoT). And according to a Moody's report, a single IT security firm reported a nearly 10,000% increase in attempted attacks on healthcare customers in 2020 over the prior year.
Many devices in operation today were not built with cybersecurity risks in mind, leaving many devices vulnerable to attack. Especially those with outdated hardware, software and protocols. As a result, the FDA has emphasized the need for a total lifecycle approach to managing cybersecurity risks in new medical devices and also that such risks should be considered early in the device design and approval process.
Regulations addressing cybersecurity risks in new medical device development will undoubtedly proliferate and become more complex in the months ahead. But given the speed of technological innovation and attendant threats, regulators are in a constant race to provide relevant and responsive guidance. That means figuring out what it takes to pass FDA muster will become increasingly challenging amidst a complex and already burdensome approval process.
Here's what MedTech and life sciences companies need to know.
How does the FDA regulate cybersecurity in medical devices?
While the vulnerability of particular sectors, like healthcare, to cybersecurity threats has been magnified by the recent proliferation of ransomware and other cyberattacks, there is surprisingly no current statutory requirement in the U.S. that expressly requires medical device manufacturers to address cybersecurity in the pre or postmarket review process.
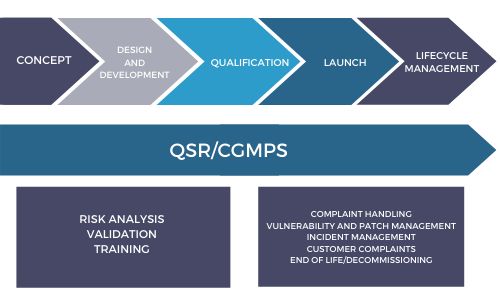
The FDA currently addresses cybersecurity in its Current Good Manufacturing Practices (CGMPs) as codified in the Quality System Regulations (QSR). The QSRs are designed to ensure that medical devices are manufactured, maintained, and quality issues monitored and corrected to ensure their continued safety and effectiveness for end-users throughout a product's life cycle.
Per QSR requirements, medical device manufacturers in the U.S. or those importing devices for use in the U.S. must demonstrate adequate cybersecurity design controls as part of the premarket application (PMA) and 510(k) submission process. The FDA has made recent moves to harmonize QSR requirements with international standards, such as ISO 13485, as are reflected in its current draft guidance on the consideration of cybersecurity in medical devices in the premarket submission process.
This draft guidance outlines key elements that should be considered in the device design and development process, including:
- "identification of assets, threats, and vulnerabilities;
- assessment of the impact of threats and vulnerabilities on device functionality and end users/patients;
- assessment of the likelihood of a threat and of a vulnerability being exploited;
- determination of risk levels and suitable mitigation strategies; and
- assessment of residual risk and risk acceptance criteria."
Cybersecurity risks should be addressed early in the medical device design process
The Department of Health and Human Services (under which the FDA operates) in its fiscal year 2021 Congressional budget justification indicated that as part of the pre-market approval process, new medical device manufacturers will be required to submit evidence of cybersecurity capabilities built into their device design and architecture.
According to Kevin Fu, the FDA's Center for Devices and Radiological Health's newly appointed acting director of cybersecurity, having robust threat models in the early design process is critical to mitigating the risks of ransomware and other external threats.
"Ransomware is a symptom of shortcomings in threat models during early medical device design."
"A medical device with an appropriate threat model for these foreseeable risks can then deliberately include design controls
to withstand the deleterious effects of ransomware."
-Kevin Fu, CDRH Acting Director Medical Device Cybersecurity
The FDA's guidance on cybersecurity considerations in the premarket submission process also emphasizes the need to consider cybersecurity risks early in the design and development process. Revealing potential cybersecurity problems early in the device design process can help smooth the pre-market approval process and avoid the additional costs and delays of taking a design back to the drawing board.
Design controls should be tailored to the risk
Because the QSRs cover a broad range of devices, from pacemakers to simple surgical tools, they are intentionally designed as a flexible framework rather than a prescriptive set of rigid requirements.
As a result, while the FDA has defined the essential elements that every quality system should embody, it is up to the individual manufacturer to determine the appropriate procedures and processes given the manufacturer's resources, the relative risks, and the necessity for each QSR element.
The International Medical Device Regulators Forum's (IMDRF) Principles and Practices for Medical Device Cybersecurity outlines a helpful approach to analyzing cybersecurity risks in the medical device design process. (The IMDRF is a global task force of medical device regulators, including the FDA, that is currently working together to accelerate the implementation of an international medical device regulatory scheme).
The IMDRF emphasizes that risk analyses should focus on the risk of patient harm, which is impacted by both:
(1) the exploitability of vulnerabilities, and
(2) the severity of harm to patients if a vulnerability is exploited.
In line with this approach, the extent of cybersecurity design controls needed for a particular device will depend on how the device will be used, the relative risks and vulnerabilities, and the environment in which it will be used.
For example, devices that are connected to other products, networks, or the internet will require more extensive design controls than standalone devices with no connection. Likewise, certain devices, such as pacemakers and brain stimulators present a more serious risk of patient harm in the event of a cybersecurity breach than diagnostic or imaging devices.
Quality design is all in the process
One mistake that medical device innovators often make is thinking of QSR compliance as a "check the box" activity that can be done ad hoc and after the fact. But "quality" is not something that is simply added in at the end of the device design process like a sprinkle of salt and pepper.
The QSRs are designed precisely to ensure that the development and design process is embedded with quality system principles that result in a safe and effective product. This means that when it comes to cybersecurity, medical device developers must think early about the product's total lifecycle, the vulnerabilities and risks at each stage of the lifecycle, and how their product is designed to mitigate those risks. The best way to do that is to start early and build these considerations into the process.
Effective cybersecurity must be integrated into the quality system process from the outset. Incorporating quality system principles early in the design process will ensure a disciplined approach to design validation and documentation that ensures cybersecurity vulnerabilities and the impacts of their exploit are mitigated throughout the product's entire lifecycle.
Having a regulatory/quality systems expert on your team is critical to premarket approval success
In my many years of experience advising medical device companies, one of the top causes for serious problems from an operational or regulatory perspective is not having a multi-talented team that includes an expert in quality systems regulation and process design. This is no less true, and perhaps even more important when it comes to designing a new medical device to withstand cybersecurity risks.
The FDA's rules and process for considering cybersecurity risks in the premarket approval process are rapidly evolving to meet new threats. Medical device sponsors need a trusted advisor who can help them stay in step.
Crowley Law LLC is a full-service law firm for tech and life science companies. With decades of experience in the tech and life-sciences space, our firm pulls from a deep network of consultants and technical experts who can help you navigate the regulatory hurdles that are inherent to new device development.
Cybersecurity is just one of many issues important for new medical device manufacturers to consider early in the design and development process. As a boutique law firm Crowley Law LLC specializes in delivering targeted legal advice that meets your company's specific needs.
Originally published 9.1.21
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
