
- within Food, Drugs, Healthcare and Life Sciences topic(s)
- within Food, Drugs, Healthcare, Life Sciences, Technology and Insurance topic(s)
Since 2013, the National Medical Products Administration ("NMPA") Center for Drug Evaluation ("CDE") has been releasing a Drug Review Annual Report ("Report") each year, which summarizes its work on drug review of the previous year. The Reports have gradually covered key issues such as the acceptance of drug registration applications, the review and approval of drug registration applications, the expediting procedures, the communication mechanism, main issues on drug registration applications, the varieties in key therapeutic areas, and the review and approval of traditional Chinese medicines.
We have made a comprehensive analysis of the latest seven Reports (from 2015 to 2021), in order to provide an overview of the progress of CDE's work regarding drug registration and show the past achievements and future development trends in China's reform of the drug review and approval system. We hope this will provide some guidance for future drug development and registration activities.
Highlights of China's reform of the drug review and approval system
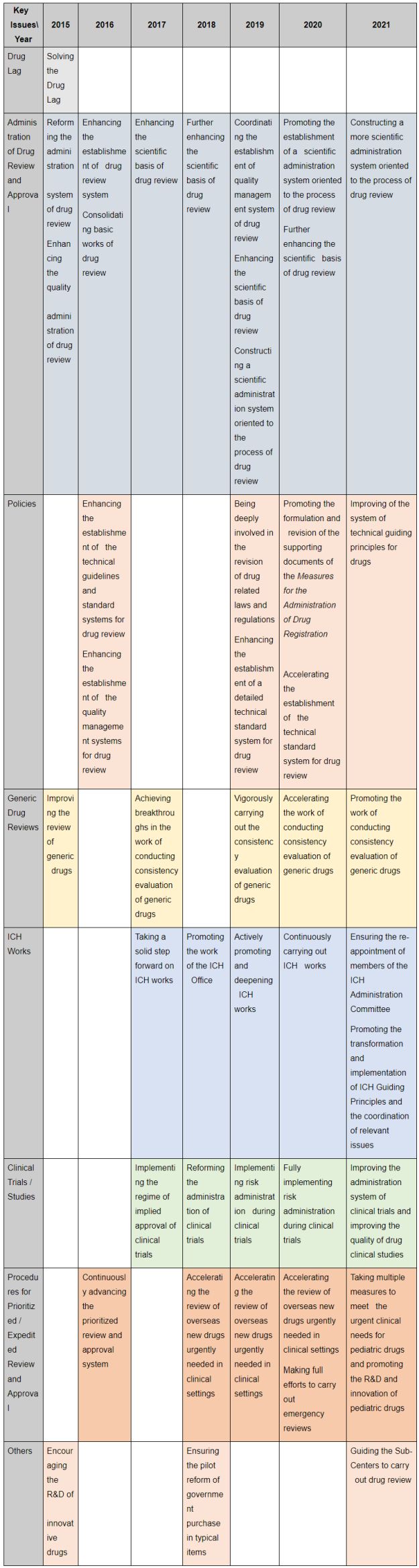
Each year, CDE lists and explains in the Reports its main work and progress regarding drug review. Therefore, we can find out the priorities of CDE's work and the focus of the reform of China's drug review and approval system for each year.
The following table extracts part of the keywords of the reform mentioned in the Reports over the past seven years. Among them, addressing the drug lag issue was the focus in 2015, while generic drug review (5 hits), ICH works (5 hits), clinical trial administration (5 hits), and procedures for prioritized/expedited review and approval (5 hits) were the keywords repeatedly mentioned in the past six years. According to these highlights, we summarize the development of China's drug review and approval system in the past seven years covering five topics including,
1. the time management of drug review and approval,
2. the modification in the registration classification and in the application requirements of chemical drugs,
3. the consistency evaluation of generic drugs,
4. the development of the registration of biologics, and
5. the review and approval of pediatric drugs and orphan drugs.
Main issues on drug registration applications
In the 2020 Report and the 2021 Report, CDE has respectively listed and analyzed the main issues on drug registration applications of the year. This article will summarize these main issues in order to offer some help and suggestions for those participating in the R&D and the registration of drugs.
I. Main issues and analyses
The following table summarizes the main perspectives and specific issues in drug registration applications:
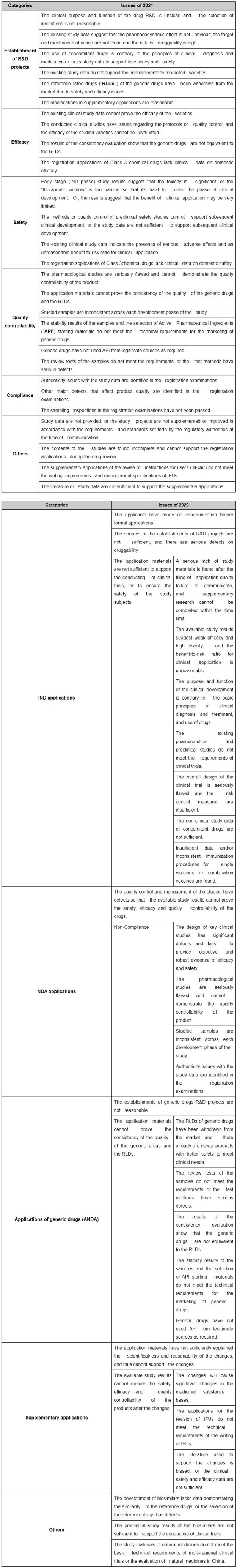
According to the data provided in the 2021 Report, 542 applications were concluded as disapproved/recommended for disapproval after technical review, of which 66.3% were due to the applicants' failure to supplement materials within time limit, and 33.7% were due to the failure of application materials to prove the safety, efficacy or quality of the drugs. CDE has pointed out that, overall, the main issues in the registration applications in 2021 have great similarity with that in the previous years, but there are also some changes that deserve attention:
Firstly, new issues that emerged in 2021 include the failure of applicants to submit study materials as requested by the regulatory authorities during pre-IND communications, resulting in the lack of study materials during the review period of IND applications. According to Article 88 of the current Measures for the Administration of Drug Registration, the applicant shall not submit supplementary technical materials during the review period for an application for clinical trials or a supplementary application during the clinical trials. Therefore, if the applicant fails to submit the study materials that have been requested by the regulatory authorities during the communications, the application will not be approved.
Secondly, the 2020 Report has listed 12 main issues and 19 specific issues, while the 2021 Report has listed 28 main issues but no specific issues. Although the classification of issues in the 2020 Report and the 2021 Report are not the same, CDE has implied that the number of drug varieties rejected due to certain reasons in 2021 has changed compared with in 2020: (i) there were no application rejected due to lack of communication in 2021; (ii) the number of applications of Class 3 chemical drugs rejected due to lack of clinical data on domestic efficacy and safety has increased significantly in 2021 compared to previous years; (iii) the number of applications rejected due to compliance issues was decreasing in 2021 compared to previous years; and (iv) the number of applications rejected due to the reasonability of the establishment of R&D projects has increased significantly in 2021.
II. Insights and suggestions
With reference to CDE's analysis of the aforementioned issues, participants in drug R&D and registration may consider the following recommendations:
Firstly, need to pay attention to the basis of drug R&D projects. In recent years, as more attention is paid to the regulatory compliance issues related to drug registration applications, the non-compliance issues have been relatively reduced. However, beside red lines, the reasonability of the drug R&D projects must also be taken seriously as CDE has been raising the quality requirements for drug R&D. The Reports emphasize that the R&D of drugs should be based on clinical needs, and should especially aim at fulfilling those unmet clinical needs. The R&D of drugs should be oriented by clinical value and should avoid low-level or repetitive development. The clinical value and advantages of Class 2 chemical drugs should be fully evaluated.
Secondly, the communication mechanism should be properly utilized. Relevant parties should be aware that the communication mechanism not only offers important facilities for registration applications, but also may provide specific regulatory requirements to registration applications. On one hand, the applicants can make full use of the communication mechanism to strengthen their communication with regulatory authorities in all aspects of drug development, so to eliminate information asymmetry and reach consensus. On the other hand, the applicants must pay attention to and follow the requirements provided by regulatory authorities during the communication; otherwise, the registration applications may fail.
Thirdly, updates of the filing requirements for drug registration made by regulatory authorities should be timely followed. As mentioned above, after the implementation of the current Measures for the Administration of Drug Registration, the requirements for the registration applications of Class 3 chemical drugs have been adjusted, and the 2020 Clinical Technical Requirements for Drugs Marketed Overseas but Not Marketed in China clearly stipulates the requirements of the clinical trials for the registration of Class 3 chemical drugs. Understanding and following these latest regulatory requirements helps to avoid issues such as the failure of registration of Class 3 chemical drugs for lack of clinical data on domestic efficacy and safety.
Finally, basic research of innovative drug development in the early stage should be strengthened. The Reports have mentioned that it is advisable to do sufficient assessment on the druggability for certain new mechanisms of action and new targets and to carry out as many proof-of-concept studies as possible to reduce the risk of subsequent development, so that the waste of research resources can be avoided.
Time management of drug review and approval
I. Drug Lags and its causes
The backlog of drug review and approval ("Drug Lags") has long been a serious issue for China's drug administration department. The 2015 Report clearly states that "the large amount of backlog of review tasks has become a huge obstacle to the achievement of scientific supervision and healthy development of the industry". At its peak in 2015, CDE had a backlog of over 22,000 review tasks. Therefore, it has become an arduous task for the authorities to solve the Drug Lag issue.
There are many reasons for the Drug Lags: First, there is an unbalance between the review workload and the review capacity. After the year of 2000, the authority of drug review and approval was taken over by the central government. As a result, review and approval tasks that were originally scattered in the provincial governments become centralized. The huge increase in review workload severely challenges the meager resources of drug review. Second, review timelines are lengthy and continually delayed. According to statistics, between the year of 2013 and 2015, the average delay period for the review and approval of the applications for innovative drug clinical trials in China is 14 months1. Finally, serious problems of poor-quality and duplicate fillings and incomplete, untrue and non-standardized filling materials have further led to the Drug Lags.
II. Main countermeasures
According to the Opinions of the State Council on Reform of the Review and Approval System of Drugs and Medical Devices ("Notion No.44"), settlement of Drug Lags is one of the primary goals of the system reform. Notion No.44 proposes to "clear the backlog by the end of 2016, achieve yearly balance between registration applications and reviewed cases as soon as possible, and reach the goal of reviewing and approving in accordance with prescribed time limit by 2018."
In order to solve the Drug Lags, China has adopted, including but not limited to, the following measures in recent years:
First, to conduct self-examination and verification of drug clinical trial data. As mentioned above, untrue and non-standardized filings is one of the reasons for the Drug Lags. To this end, the former State Food and Drug Administration of China ("CFDA") issued the Notice on the Self-examination and Verification of Drug Clinical Trial Data ("the Notice") on July 22, 2015, to verify the clinical trial data of received registration application for drugs that are applying for manufacturing or importation. The Notice requires 1,622 clinical trial projects to conduct self-examination and voluntarily withdraw itself in case of any problem discovered. If failing to submit a report or withdraw the application within prescribed time limit, unannounced inspections would be conducted. Once any problem were discovered, the relevant applicant, clinical trial institution and personnel related would face serious administrative penalties. As of December 31, 2015, 1,009 drug registration applications had been voluntarily withdrawn by the applicants, representing 62.2% of the total drug registration applications, involving hundreds of drug companies. Subsequently, Working Procedures for Drug Clinical Trial Data Verification (Interim), Drug Administration Law, Measures for the Administration of Drug Registration, Start-up Working Procedures for Verification and Inspection of Drug Registration, Working Procedures for Drug Registration Verification (for Trial Implementation), Key Points and Principles for Drug Registration Verification (Drug Clinical Trial) (for Trial Implementation), and other relevant regulations were issued one after another. Self-examination and verification of drug clinical trial data changes from an unexpected regulatory storm to a sophisticated, long-term regulatory system. Implementing the self-examination and verification of drug clinical trial data has both decreased the quantity of low-quality registration applications and improved the overall quality of registration applications, significantly assisting in the reduction of application backlog.
Second, to establish the implied approval policy for clinical trials. As mentioned above, the delay of review and approval, especially in clinical trial application is one of the reasons for the Drug Lags. In this regard, Opinions on Deepening the Reform of the Review and Approval Systems and Encouraging Innovation on Drugs and Medical Devices proposes to optimize the approval procedures for clinical trial application. Accordingly, drug administration department shall be deemed to have approved if it does not give a denial or doubting opinions within a given period after accepting an application for clinical trial, and the registration applicant may conduct the clinical trial in accordance with the submitted protocol. Later in 2019, Article 19 of the revised Drug Administration Law formally established the implied approval policy for clinical trial applications in China, significantly shortening the review and approval time limit to 60 working days. This greatly accelerates the review and approval process of drug clinical trials in China.
Third, the establishment of expediting registration procedures for drug marketing. Chapter IV of the Administrative Measures for Drug Registration revised in 2020 provides principal provisions on the procedures for breakthrough therapy designation, conditional approval, prioritized review and approval and procedures for special approval. The follow-up legislations including the Working Procedures for Breakthrough Therapy Designation (for Trial Implementation), Working Procedures for the Review and Approval Procedures for Conditional Approval of Drug Marketing Applications (for Trial Implementation) and the Working Procedures for the Prioritized Review and Approval Procedures for Drug Marketing Applications (for Trial Implementation) further promote the implementation of expediting registration procedures.
Fourth, the expansion of the review capacity. The 2019 Report proposes "to expand review capacity through multiple channels including but not limited to large-scale recruitment of personnel and secondment of provincial bureau personnel". Later in 2021, the Report emphasizes to promote team building by "actively coordinating and increasing staffing, promoting the recruitment of talent based on review needs, and reinforcing the professional review capacity". It is thus clear that promoting team building has always been the focus of CDE in recent years.
III. Achievements and progress
According to the data from annual Reports, the Drug Lags was progressively reduced from 2015 to 2018. In 2015, the number of applications for review was reduced from more than 22,000 at the peak to less than 17,000 at the end of the year; in 2016, the number of applications for review dropped to nearly 8,200; In 2017, the number of applications for review dropped to 4,000, basically reaching the goal of eliminating the Drug Lags stipulated in Notion No.44. In 2018, the number of applications for review dropped to 3,440, further consolidating the requirement of Notion No.44.
The 2019 Report points out that the Drug Lags was basically eliminated in 2019, and the work focus of CDE has gradually shifted from resolving Drug Lags to improving the percentage of drug registration application received and approved in time ("Timeliness Percentage"). According to the data annually published by CDE, the overall Timeliness Percentage throughout the whole year increased year by year from 2019 to 2021. Timeliness Percentage for traditional Chinese medicine, chemical medicine and biological products reached 90% in 2019, 94.48% in 2020 and 98.93% in 2021.
Click here to continue reading . . .
Footnotes
* Leyi Wang and Shuwen Sun have also contributed to this article.
1. Zhou Q, Chen XY, Yang ZM, Wu YL (2017), The Changing Landscape of Clinical Trial Approval Processes in China, Nature Reviews Clinical Oncology 14:577–583.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
[View Source]

