

- with readers working within the Aerospace & Defence and Retail & Leisure industries
- within Finance and Banking, Law Department Performance and International Law topic(s)
Background
The medical device industry in India has witnessed sweeping changes in the last five years, especially on the regulatory front. To begin with, the Medical Devices Rules, 2017 was enacted inter alia to regulate and standardize medical devices in India, w.e.f. January 1, 2018. This was soon followed by two notifications of the same date, i.e., February 11, 2020 from the Ministry of Health and Family Welfare (MoHFW) which in one hand expanded the scope of medical device and on the other, mandated importers and manufacturers of specified medical devices to obtain registration and import license within a defined timeline. These steps seem to be a way forward to regulate and standardize the medical device industry with uniformed quality standards.
We have summarised below the relevant laws and its requirements, with focus on registration for importers of medical devices.
Applicable Laws and Analysis

Scope of Medical Devices Widened
The MD Rules inter alia provides for the regulation, manufacture, import, labelling and sale of medical devices in India, with effect from January 1, 2018. These MD Rules inter alia are applicable to all medical devices notified from time to time under Section 3 (b) (iv) of the D&C Act. Section 3 (b) (iv) of the D&C Act provides for the definition of drug vis-à-vis medical device.
Subsequently, the MoHFW vide notification dated February 11, 2020 [S.O. 648 (E)] notified the following as 'medical devices' under Section 3(b)(iv) of the D&C Act ("Newly Notified Devices") w.e.f. April 1, 2020 by further broadening the definition of a medical device:
"All devices including an instrument, apparatus, appliance, implant, material or other article, whether used alone or in combination, including a software or an accessory, intended by its manufacturer to be used specially for human beings or animals, which does not achieve the primary intended action in or on human body or animals by any pharmacological or immunological or metabolic means, but which may assist in its intended function by such means for one or more of the specific purposes of ? (i) diagnosis,prevention, monitoring, treatment or alleviation of any disease or disorder; (ii) diagnosis, monitoring, treatment, alleviation or assistance for, any injury or disability; (iii) investigation, replacement or modification or support of the anatomy or of a physiological process; (iv) supporting or sustaining life; (v) disinfection of medical devices; and (vi) control of conception."
The intention behind the notification of Newly Notified Devices is that if a device, including software or accessory, is intended by its manufacturer to be used for assisting in any of the above specified purpose, then it would fall within the ambit of "medical device" and be covered under the MD Rules. This brought several devices under the scope of MD Rules, due to the intended purpose (especially on account of the wide range of diagnosis, monitoring, treatment), although such devices may not be per se a 'medical' device.
New Requirement of Registration of Medical Device
The MoHFW on the same day of notification of Newly Notified Devices i.e., on February 11, 2020, also notified the Medical Devices (Amendment) Rules, 2020 ("Registration Rules"), which is applicable to all medical devices notified under Section 3 (b) (iv) of the D&C Act (and thus includes Newly Notified Devices), except the 37 devices specified in the Annexure of Eighth Schedule of the Registration Rules.
Manufacturers and Importers of such medical device had to voluntarily register their medical devices with the Drug Controller General of India through the online portal viz. Online System of Medical Devices, before October 1, 2021; post which registration was made mandatory.
Further on a side note, the Registration Rules also mandates importers and manufacturers of specified medical devices to obtain license to import by August 10, 2022 (Class A and Class B devices) and August 10, 2023 (Class C and Class D devices). Though it has not been specified, however if registration and/or import license is not obtained by the given dates, then it may amount to contravention of the D&C Act/MD Rules and manufacturers and/or importers, unless said dates are extended, may not inter alia be able to sell or import the medical devices until registration or import license is obtained.
We provide below a snapshot of the voluntary registration process for an importer.
Step 1: Register (create account) on Online System of Medical Device
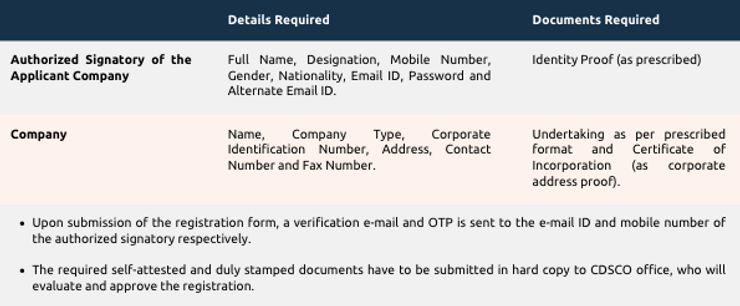
Step 2: Registration on Online System of Medical Device
- Undertaking in terms of Rule 19 D (2) (v) of the Registration Rules, signed by the Authorized Signatory has to be uploaded.
- Manufacturing site details, including but not limited to, manufacturer name, address, site type and contact details have to be provided.
- Device details, including but not limited to, purpose, category, grouping description, notified category of medical device, dimension, model details, accessories/component details, polymer coating details have to be provided along with submission of Free Sale Certificate and Certificate of Compliance with respect to ISO 13485.
Once the above information is submitted, a file number is generated immediately which is the registration number of the applicant company. The said registration number has to be compulsorily declared on the product label w.e.f. June 1, 2022 in terms of the recently notified Medical Devices (Amendment) Rules, 2022 dated January 18, 2022.
Conclusion
The enforcement and insistence on registration of medical devices and obtaining import license have gained momentum in the last few months. As per recent media report, Food and Drugs Administration, Maharashtra has issued letters to all medical device manufacturers to register themselves and obtain a license within the stipulated timeframe, failing which they will face action for violation under applicable laws.
Although the industry is gearing up for registration, it is a fact that complete preparedness and readiness are yet to be achieved inter alia due to the requirement of submission of ISO 13485 compliance certificate and Free Sale Certificate. Various representations have been received by the MoHFW from industry bodies in this regard, which is currently under consideration.
The enforcement of these regulatory framework will definitely set a quality benchmark of medical devices and ensure that superior and standardized quality medical devices are provided to patients, but the actual implementation may take some time.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.

