

Key Takeaways:
- The Food and Drug Administration (FDA) provided draft guidance on its expectations surrounding Predetermined Change Control Protocols (PCCP), which would be submitted as part of a premarket submission filing for certain software-related medical devices that incorporate machine learning technology.
- A PCCP allows the FDA to pre-authorize certain anticipated changes to the software that will not need a new submission when they are made in accordance with a modification protocol.
- Modifications to a device that would result in a new intended use or indications for use would not likely be appropriate for a PCCP, and modifications to a PCCP would likely require a new submission.
On April 3, 2023, the Food and Drug Administration (FDA) published in the Federal Register an announcement of availability for the draft guidance, Marketing Submission Recommendations for a Predetermined Change Control Plan for Artificial Intelligence/Machine Learning (AI/ML)-Enabled Device Software Functions (April 3, 2023). The draft guidance would define and implement processes for the FDA's review and authorization of Predetermined Change Control Protocols (PCCP) for modifications to machine learning used by marketed medical devices, allowing certain such modifications to occur without a new premarket submission to the FDA. Because the policies in the draft guidance would apply only to the category of artificial intelligence known as machine learning (ML), this alert will refer to ML and will refer to the draft guidance as the "Draft ML PCCP Guidance."
Background
Medical devices that rely on ML include devices with embedded ML software (Software in a Medical Device or SIMD) and ML software that is the device (Software as a Medical Device or SaMD). Because software is iterative and evolves rapidly, SiMD and SaMD present unique challenges to the FDA's traditional authorities for medical devices. SiMD and SaMD that use ML may evolve automatically in ways that are nontransparent to the user, amplifying these challenges.
At least since the publication of the Proposed Regulatory Framework for Modifications to Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device (SaMD) Discussion Paper and Request for Feedback ("2019 discussion paper"), the FDA has signaled that it intends to rely on PCCPs to address the challenge of ML-enabled devices. The FDA affirmed this intent in 2021 with its publication of Artificial Intelligence/Machine Learning (AI/ML)-Based Software as a Medical Device Action Plan ("the Action Plan"). The FDA gained express PCCP authority with passage of the FDA Omnibus Reauthorization Act (FDORA) in December 2022. The Draft ML PCCP Guidance describes how manufacturers can use PCCPs for ML-enabled devices. Specifically, the Draft ML PCCP Guidance (1) defines relevant terms; (2) describes a proposed policy for submitting, relying on, and modifying a PCCP, including broad categories of modifications appropriate for a PCCP; and (3) identifies the contents of a PCCP. The guidance also provides a model for developing the Modification Protocol of the PCCP and provides several hypotheticals to illustrate the proposed policy.
Terminology
The Draft ML PCCP Guidance introduces several new terms and regulatory concepts:
- Device Software Function (DSF)/Machine Learning-Enabled Device Software Function (ML-DSF): The Guidance clarifies that a device software function is a distinct purpose of the software, which could be the intended use or a subset of the intended use of the product. The ML-DSF is an ML-enabled device software function.
- Data Sets: The Draft ML PCCP Guidance defines three categories of data used to develop, evaluate, and test MS-DSFs: training, tuning, and testing data sets. Training and tuning data are used in subsequent phases of developing an ML algorithm. The FDA notes that these "data typically should be representative of the proposed intended use populations (e.g., with respect to race, ethnicity, disease severity, gender, age, etc.)." Testing data is the data used in testing to establish a reasonable assurance of safety and effectiveness in a premarket submission. Importantly, the guidance notes that "[t]esting data should be independent of data used for training and tuning and should be from multiple sites different from those that were used to generate training."
- Predetermined Change Control Plan/Authorized Predetermined Change Control Plan (PCCP): The guidance defines a PCCP as "documentation describing what modifications will be made to the ML-DSF and how the modifications will be assessed. The modifications described in the PCCP include device changes that would otherwise require a PMA supplement, de novo submission, or new 510(k) notification." PCCPs include a Description of Modifications, a Modification Protocol, and an Impact Assessment, which are described below. An authorized PCCP is a PCCP that has received FDA authorization.
Policy: FDA Authorization of a PCCP Allows Covered Modifications without FDA Review
After the FDA clears a premarket notification ("510(k)") submission, grants a petition for de novo review, or approves a PreMarket Approval (PMA) application, certain modifications to the device may trigger the need for a new premarket submission. Established policies in FDA regulations and guidance documents describe when post-market modifications to a device require a new submission, including 21 C.F.R. 807.81(a)(3) and Deciding When to Submit a 510(k) for a Software Change to an Existing Device for 510(k)s and 21 C.F.R. 813.29(a) and Deciding When to Submit a 510(k) for a Change to an Existing Device or Modifications to Devices Subject to Premarket Approval (PMA) - The PMA Supplement Decision-Making Process for PMAs. The Draft ML PCCP Guidance relies on principles in these regulations and guidance documents to define the modifications that a PCCP should address and should be read in conjunction with them.
As specified in FDORA, the Draft ML Guidance states that PCCPs must be submitted as a part of a premarket submission and recommends that the PCCP constitute a clearly identified standalone section of the submission. The PCCP should also be referenced elsewhere in the submission, including in the device description, labeling, and relevant sections used for determining substantial equivalence or reasonable assurance of safety and effectiveness. The draft guidance notes that for 510(k) submissions, the FDA will determine substantial equivalence by comparing the device under review to the version of the predicate device cleared or approved prior to any changes made under the PCCP. The guidance describes an authorized PCCP as a technological characteristic of the device, meaning changes to the PCCP will require review under the same principles that apply to other device technological characteristics.
Once the PCCP is authorized by the FDA, manufacturers should assess any modifications to the device to determine whether they are (1) specified in the authorized PCCP and (2) implemented in accordance with the Modification Protocol of the authorized PCCP. If the answer to these questions is yes, no premarket submission concerning the change is required; the manufacturer should document the change in accordance with the Quality System regulation, 21 C.F.R. Part 820. The Draft ML PCCP Guidance provides a flow chart for manufacturers with an authorized PCCP to follow to determine whether a device modification requires a new submission.
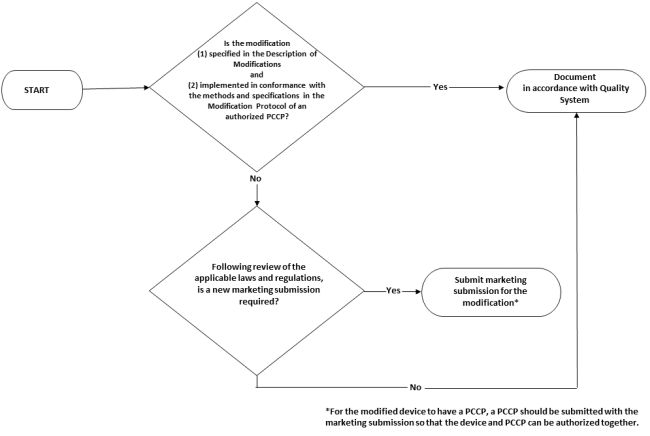
If the answer to either question is no, however, the manufacturer must follow the usual process for determining whether the modification triggers the need for a new premarket submission. For example, if the manufacturer plans to modify a 510(k) cleared ML-enabled device in a way that is not described in the authorized PCCP, the manufacturer should evaluate the modification in accordance with 21 C.F.R. 807.81(a)(3) and the guidance document, Deciding When to Submit a 510(k) for a Software Change to an Existing Device.
A new marketing submission may be required for a device modification effected through a change to the authorized PCCP or a device modification that is outside the authorized PCCP. Either way, if a new marketing submission is required, the manufacturer should include a PCCP, modified, as necessary, for the modified device.
Moreover, if a manufacturer seeks to make modifications to the authorized PCCP, the FDA expects such modifications will themselves generally constitute changes that require a new marketing submission for the device because a modification to the PCCP will generally significantly affect the safety or effectiveness of the device.
Modifications Appropriate for a PCCP
The Draft ML PCCP Guidance notes that only certain modifications are appropriate for a PCCP, and identifies the following types of modifications as ones that "may be acceptable":
- modifications related to quantitative measures of ML performance (such as improvements to performance resulting from re-training the ML model with new datasets from the same intended use population);
- modifications related to inputs to the ML-DSF (such as expanding the algorithm to accept input from different makes, models, or versions of a data acquisition system) or limited modifications related to new types of inputs; and
- limited modifications related to the device's use and performance (such as authorization of a device for a subset of a population within the originally indicated population).
Despite this third category of changes acceptable for a PCCP, the Guidance notes that modifications that create a new intended use, are not appropriate for a PCCP. In addition, the Guidance states that "[a]t this time" the FDA does not expect that modifications that create a new indication for use will be appropriate for a PCCP. This exclusion seems likely to elicit comment from manufacturers of ML-enabled devices seeking to use a PCCP to expand the indications for use without the need for a new product submission—such manufacturers will need to convince the FDA that changes to indications for use can be reviewed in a PCCP in a way that ensures continued substantial equivalence to the predicate or provides a continued reasonable assurance of safety and effectiveness.
The Draft ML PCCP Guidance also distinguishes between changes to ML-DSFs that occur automatically—meaning the algorithm evolves without additional human intervention—and those implemented manually—meaning the changes require "human input, action, review, and/or decision-making"—and notes that the FDA is proposing to allow PCCPs for both. Concerning automatically implemented changes, however, the FDA notes the additional complexity, and emphasizes the importance of the benefit-risk assessment in evaluating such changes.
Components of the PCCP
As noted in the Terminology section of this alert, a PCCP has the following three sections:
Description of Modifications: While the PCCP may include multiple modifications, the FDA recommends that the PCCP limit the number to specific, planned modifications that can be verified and validated in accordance with the Modification Protocol. The Description of Modifications should include the specific rationale for the planned change to each part of the ML-DSF. The FDA may authorize some, but not all planned modifications.
Modification Protocol: The Modification Protocol should describe the methods to be used to develop, validate, and implement modifications identified in the Description of Modifications. The Modification Protocol should include: 1) data management practices; 2) re-training practices; 3) performance evaluation protocols; and 4) update procedures, including communication to users, for each modification in a PCCP. The protocol should show that the methods described will ensure the safety and effectiveness of the modified device and should follow 21 C.F.R. Part 820, including Quality System requirements for design, product, and document controls.
Impact Assessment: The Impact Assessment documents benefits, risks and any mitigations of implementing a PCCP for an ML-DSF in accordance with the manufacturer's Quality System.
Stakeholders have 90 days to submit comments on the Draft ML PCCP Guidance, which the FDA will consider before issuing a final guidance.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.

