
The medical device segment in India, considered Asia's fourth largest market worth approximately USD 5.5 billion and expanding at a steady pace, presents an exciting business landscape and opportunities for both domestic as well as international manufacturers/entrepreneurs. At present, India's medical device sector is dominated by multinational companies, which is evident from the fact that India relies on imports of medical devices (about 75-80% of the sales are generated by imported medical devices) for supply to its healthcare system. Over the years, many multi-nationals have set up operations in India.
On August 06, 2018, the Indian Pharmacopoeia Commission (IPC), the National Coordinating Centre (NCC) of Materiovigilance Programme of India (MvPI) released Draft Guidance Document for Medical Devices1 to provide necessary information to all the stakeholders of the country regarding the regulatory requirements, quality management systems and standards required to be followed for medical devices. The new draft includes standards for medical devices as a Guidance Document for the benefit of the general public, patients and healthcare professionals. It will also serve as a reference manual for the licensing authority in the matters relating to medical devices. The guideline also describes post-market requirements for medical devices.
However, the nature of majority of the operations in India is to only distribute imported devices and provide support function. From this view, the purpose of this document:
- Aims to be informative in nature on medical device standards comprehensively, irrespective of usage i.e. whether on human beings and animals. Preparation of standards on medical devices nationally and internationally is an ongoing process, irrespective of regulation on same by National/State Medical Devices Regulator.
- Provide guidance to assist manufacturers, traders/distributors, importers, clinical establishments, healthcare professionals and general public on nationally-recognized medical devices standards and other regulatory requirements concerning medical devices in India.
- Serves as ready reference for medical devices standards preparation/adoptions, clinical care quality bodies, nomenclature of medical devices, claims on medical devices and validation mechanism, existence of multiple regulatory bodies on medical devices and law (ACT) directly governing medical devices in India.
Medical device Regulation in India
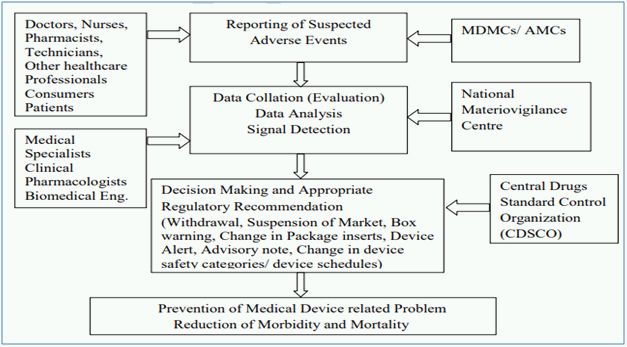
The Central Drugs Standard Control Organization (CDSCO) under Directorate General of Health Services in Ministry of Health and Family Welfare (MoHFW), Government of India (GoI), is the National Regulatory Authority (NRA) responsible for approval of manufacturing, import, conduct of clinical trials, laying down standards, sale and distribution of medical devices through enforcement and implementation of the Medical Devices Rules, 2017, released dated 31st January 2017 by the MoHFW. As NRA, CDSCO has the responsibility to conduct MvPI wherein, Indian Pharmacopoeia Commission functions as NCC for MvPI. MvPI is meant to enable safety data collection in a systematic manner so that regulatory decisions and recommendations on safe use of medical devices being used in India can be based on data generated here. The programme is meant to monitor medical device-associated adverse events (MDAE), create awareness among healthcare professionals about the importance of MDAE reporting in India and to monitor the benefit-risk profile of medical devices.
The draft guidance for medical device covers the regulatory approval process, classifications and grouping methodology, quality management system, labelling requirements including post marketing surveillance as draft highlights described below:
Classification of medical devices
The draft guidance describes the basic principles and parameters involved in classification of medical devices & in vitro diagnostic medical devices (hereafter referred as IVD medical device) based on the below facts:
- Classification of Medical devices based on the risk parameters as described, namely: (i) Low risk - Class A; (ii) Low moderate risk- Class B; (iii) Moderate high risk- Class C; (iv) High risk- Class D.
- Classification of Medical devices and in-vitro diagnostic medical devices based on the intended use of the device and other parameters, where the basic principle like – Medical Device's intended purpose of use, intended to be used in combination with another device, software which drives a device or influences the use of a device, and intended to be used solely or principally in a specific part of the body are considered. Moreover, the various parameters like- invasive, non-invasive, surgically invasive and implantable nature along with the medical devices incorporating medicinal products shall also be considered during classification of devices.
However, the draft also lists medical devices and IVD medical devices class wise for reference purposes.
The essential principles of medical devices
This guidance document describes fundamental design and manufacturing requirements, referred to as "Essential Principles for Safety and Performance" that, when met, indicate a medical device including IVD medical device is safe and performs to its specification. The design and manufacturing requirements in this document are grouped in categories. This section will allow manufacturers to select the design and manufacturing requirements relevant to a particular medical device, and documenting the reasons for excluding the others.
Quality management system
This is for notified medical devices and IVD medical devices (fifth schedule of Medical Devices Rules 2017) where specific requirements for a quality management system shall be used by the manufacturer for the design and development, manufacture, packaging, labelling, testing, installation and servicing of medical devices and in-vitro diagnostics. If the manufacturer does not carry out design and development activity, the same shall be recorded in the quality management system. The manufacturer shall maintain conformity with this Schedule to reflect the exclusions.
The quality management system shall be applicable to manufacturers of finished devices, In vitro diagnostics, mechanical contraceptives (condoms, intrauterine devices and tubal rings), surgical dressings, surgical bandages, surgical staplers, surgical sutures and ligatures, blood and blood components collection bags with or without anticoagulants intended for human or animal use.
Regulatory approval process of Medical Devices in India
The guidance document describes the detailed process of registration and regulatory approval for manufacture for sale/distribution of medical devices and import of medical devices. This guidance also comprises the list of application forms and required documents and fee payable for license, permission and registration certificate to manufacture or import of medical devices. The draft also cites the audit fee payable to notified bodies registered with CDSCO to carry out audit of manufacturing sites under provisions of Medical Devices Rules, 2017.
Guidance on grouping of medical devices for product registration
The guidance explains the grouping of Medical Devices for a person who applies for license to import or manufacture for sale or distribution, sell, stock or offer for sale or distribution of medical devices as specified under respective forms to the Medical Devices Rules, 2017. The applicant may group medical devices having same or similar intended uses or commonality of technology and submit in a single application. The grouping of medical devices is for the purpose of submission of single application for license to import or manufacture in the following manner:
- Single: A single medical device is a medical device sold as a distinct packaged entity and does not meet the criteria for family, IVD test kit, system, IVD cluster or a group. It may be sold in a range of package sizes. For example - Condoms sold in package of 3, 10 or 16, can be licensed as single medical device applications.
- Family: A medical device family is a collection of medical devices and each medical device is from same license holder, is of same risk classification class, has a common intended use, has the same design and manufacturing process, and have variations that are within the scope of the permissible variants. For example - Condoms that differ in colour, size and texture but are manufactured from the same material and manufacturing process and share a common intended purpose can be licensed as a Family.
- In vitro diagnostics Test Kit: An in-vitro diagnostics kit is a device that consists of reagents or articles which are from same license holder; intended to be used in combination to complete a specific intended purpose; sold under single proprietary test kit name; and compatible when used as a test kit. For example - Human Immunodeficiency Virus (HIV) Enzyme Linked Immunosorbent Assay (ELISA) Test Kit may contain controls, calibrators, and washing buffers. All the reagents and articles are used together to detect HIV and therefore, can be licensed as Test Kit. These reagents and articles can be supplied separately as replacement items for that particular Test Kit.
- System: Medical devices comprise a system, when they are from same license holder; intended to be used in combination to complete a common intended purpose; compatible when used as system; and sold under single proprietary system name. for example - a glucose monitoring system comprising of a glucose meter, test strips, control solutions and linearity solutions can be licensed as a System.
- In vitro diagnostics cluster: An in-vitro diagnostics cluster comprises of a number of in-vitro diagnostics reagents or articles which are from same license holder; of a common methodology; sold under single proprietary name; and compatible when used as a Test Kit.
- Group: A medical device Group is a collection of two or more medical devices, supplied in a single package by same license holder, which are sold under single proprietary Group name and for a common intended purpose. For example - A first aid kit consisting of medical devices such as bandages, gauzes, drapes and thermometers, when assembled together as one package, can be licensed as a Group.
Labelling of medical devices and IVD medical devices
The guidance provides the labelling instructions of medical devices which are to be printed in indelible ink on the label, on the shelf pack of the medical device or on the outer cover of the medical device and on every outer covering in which the medical device is packed, in case of small sized medical devices on which information cannot be printed legibly, shall include the information necessary for product identification and safety. Special instructions are required if:
- Exemption of labelling requirements for export of medical devices - the labelling exemption of such device is intentional, to follow the specific requirements of law of the country to which the device is to be exported. However, the particulars like name of the device, the distinctive batch number or lot number or serial number, date of expiry, if any, the name and address of manufacturer, internationally recognized symbols in lieu of text, wherever required shall be adopted to meet the specific requirements of law of the country to which the device is to be exported.
- Unique device identification of the medical device: With effect from 1st January 2022, a medical device, approved for manufacture for sale or distribution or import, shall bear unique device identification which shall contain device identifier and production identifier.
Labelling medical device or a new in-vitro diagnostic medical device for purpose of test, evaluation, clinical investigations shall be kept in containers bearing labels, indicating the name of the product or code number, batch or lot number, serial number wherever applicable, date of manufacture, use before date, storage conditions, name and address of the manufacturer, and the purpose for which it has been manufactured.
Post market vigilance and safety requirements
MvPI has been approved since July 2015, which has Indian Pharmacopoeia Commission, Ghaziabad as the NCC for the Post market vigilance of medical devices. Presently, MvPI has 10 dedicated functional Medical Device Adverse Event Monitoring Centers (MDMCs) all over the country. All the Adverse Drugs Reaction Monitoring Centres (AMCs) under Pharmacovigilance Programme of India (PvPI) have also been entrusted to report adverse events due to the use of medical devices.
Scope of MvPI is to:
- Create a nation-wide system for patient safety monitoring.
- Analyze the benefit-risk ratio of medical devices.
- Generate evidence-based information on safety of medical devices.
- Support CDSCO in the decision-making process on use of medical devices.
- Communicate the safety information on use of medical devices to various stakeholders to minimise the risk.
- Emerge as a national centre of excellence for Materiovigilance activities.
- Collaborate with other healthcare organizations for exchange of information and data management.
The adverse event reporting system of MvPI is described below2:
Note - Guidance document for Medical Devices is neither a regulatory nor a legal document. This document has been framed on the basis of Medical Device Rules 2017, issued by Government of India. If there are any errors or omissions found in this guidance document, readers are advised to refer to original Medical Device Rules 2017. The information contained in this document should not be a substitute for Medical Device Rules 2017.
Footnotes
1 http://ipc.gov.in/images/mvpi/Guidance_Document.pdf
2 http://ipc.gov.in/images/mvpi/Guidance_Document.pdf
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.

