

Przemysł farmaceutyczny dzieli się na dwie kategorie wytwórców: firmy oryginalne i firmy generyczne.
Zazwyczaj producenci leków oryginalnych to ci, którzy najpierw poszukują nowych celów molekularnych, które mogłyby stanowić cel dla zupełnie nowych leków, a potem w toku żmudnych i kosztownych prac badawczo-rozwojowych tworzą nowe preparaty i ich formulacje, aby w toku badań klinicznych zweryfikować skuteczność danego środka i finalnie zarejestrować nowy lek.
Z kolei firmy generyczne, nazywane firmami odtwórczymi, skracają proces badawczo-rozwojowy do fazy opracowania alternatywnych formulacji lub odtworzenia kompozycji leków oryginalnych, opierając się na wynikach badań firm oryginalnych.
Coraz więcej firm generycznych rozwija segment tzw. ulepszonych generyków, czyli produktów, które nie tylko powielają znane oryginalne leki, ale też oferują wartość dodaną w postaci na przykład poprawy biodostępności czy ograniczenia skutków ubocznych albo wygodniejszej formy podawania. Choć z tego względu granice tradycyjnego podziału na leki oryginalne i generyki1 ulegają stopniowemu zatarciu, to producenci preparatów oryginalnych korzystają z zabezpieczeń prawnych dających możliwość rekompensaty kosztów prac badawczo-rozwojowych.
Skrócenie ścieżki rozwoju dla preparatów odtwórczych ma swoją cenę, firmy oryginalne korzystają z dodatkowej ochrony regulowanej w prawie farmaceutycznym. Dla farmaceutyków zarejestrowanych w procedurze centralnej jest to formuła 8+2+1, gdzie przez 8 lat nie udostępnia się wyników badań klinicznych (wyłączność danych), przez 2 kolejne lata trwa okres wyłączności rynkowej, a w przypadku zarejestrowania nowego wskazania można zyskać jeszcze dodatkowy rok ochrony. Z tego względu rozwój generycznych alternatyw leków oryginalnych jest opóźniony.
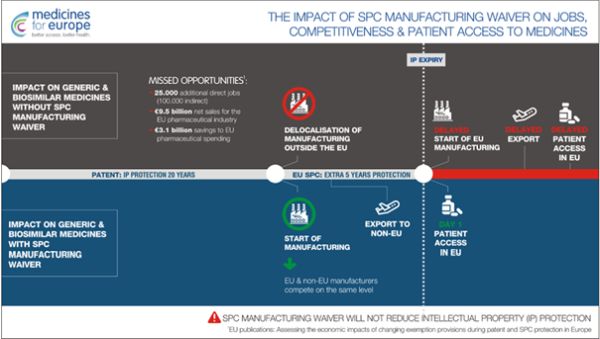
Ponadto, firmy oryginalne korzystają z ochrony patentowej dla swoich produktów zyskując 20 lat monopolu na rynku, gdzie obowiązuje patent.
Narzędziem, które pozwala na przedłużenie wyłączności korzystania z opatentowanego leku jest DPO (Dodatkowe Prawo Ochronne), powszechnie znane jako SPC (Supplementary Protection Certificate). SPC daje prawo do wydłużenia ochrony patentowej na produkt, który był przedmiotem pierwszego pozwolenia na dopuszczenie do obrotu. Na mocy regulacji unijnych rozporządzeń nr 469/2009 z 6 maja 2009 r. oraz 1768/92, wprowadzono jednolite regulacje dotyczące SPC, które umożliwiają uzyskanie dodatkowego, maksymalnie 5-letniego okresu ochrony dla produktów oryginalnych.
Zmiany rynku farmaceutycznego, zwłaszcza rosnąca w siłę pozycja firm azjatyckich powoduje osłabienie konkurencyjności firm europejskich.
W okresie trwania ochrony patentowej czy SPC, ale już po wygaśnięciu ochrony danych rejestracyjnych, firmy generyczne mogły jedynie przeprowadzić niezbędne prace badawczo-rozwojowe, które umożliwiłyby opracowanie własnego leku generycznego i przeprowadzenie procesu rejestracji.
W obecnym stanie prawnym firma generyczna może uruchomić proces produkcyjny swojego leku dopiero z chwilą wygaśnięcia SPC. Jak wiadomo produkcja leków wymaga szeregu prac przygotowawczych, zakupienia surowców, zoptymalizowania procesu wytwarzania i jego walidacji, co sprawia, że firmy oryginalne zyskują dodatkowo przynajmniej kilka miesięcy monopolu zanim producenci leków generycznych będą mogli dostarczyć swoje leki do aptek.
Ponadto wysokie koszty nowoczesnych terapii powodują znaczne ograniczenie ich stosowania, a tym samym zmniejszenie dostępności dla pacjentów. Czynniki te stały się przyczynkiem do rozważań Komisji Europejskiej na temat modyfikacji obecnego systemu ochrony patentowej w odniesieniu do SPC.
W oparciu o pogłębione analizy sytuacji rynkowej oraz wpływu na ten stan rzeczy regulacji prawnych Komisja Europejska zaproponowała nowy przywilej" dla producentów leków generycznych, tak zwany SPC Manufacturing Waiver2 (SPC MW).
Celem tej regulacji jest umożliwienie producentom
leków generycznych rozpoczęcie procesu wytwarzania
leków generycznych w okresie obowiązywania SPC, przy
czym do czasu ustania ochrony wynikającej z SPC wytworzone
preparaty generyczne mogłyby być sprzedawane tam, gdzie
ochrona SPC nie obowiązuje (na przykład do krajów
pozaunijnych). Taki przywilej" pozwalałby na wprowadzenie
do obrotu leków generycznych na rynkach europejskich
już pierwszego dnia po wygaśnięciu ochrony
SPC3. Oczekuje się, że zaproponowane
rozluźnienie ochrony wynikającej z SPC spowoduje
poprawę sytuacji firm generycznych, a pacjentom umożliwi
szybszy dostęp do tańszych odpowiedników
leków oryginalnych, dając tym samym szansę na
wcześniejsze skorzystanie
z nowoczesnych leków.
W projekcie4 zmian rozporządzenia nr 469/2009 dotyczącego SPC określono odstępstwa w ochronie produktów leczniczych objętych SPC. Oznacza to, że między innymi działania takie jak:
- wytworzenie produktu chronionego SPC lub,
- wytworzenie produktu leczniczego zawierającego ten produkt dla celu wywozu do krajów trzecich lub,
- wszelkie powiązane działania, które są bezwzględnie konieczne dla wytworzenia na terytorium Unii takiego produktu na potrzeby eksportu [na przykład import substancji aktywnej lub substancji pomocniczych, produkcja, pakowanie czy czasowe magazynowanie] lub,
- wytworzenie produktu leczniczego objętego SPC nie wcześniej niż 6 miesięcy przed wygaśnięciem SPC w celu jego przechowywania do czasu wygaśnięcia SPC, aby umożliwić jego sprzedaż po wygaśnięciu certyfikatu lub,
- przeprowadzenie wszelkich powiązanych czynności, które są bezwzględnie konieczne do wytworzenia produktu leczniczego do takiego celu nie wcześniej niż 6 miesięcy przed upływem ważności SPC.
Oprócz przywileju wynikającego z SPC MW, w celu wyeliminowania ewentualnych nadużyć oraz dla zachowania przejrzystości, do projektu zmian nr 469/2009 wprowadzono także szereg obowiązków dla producentów leków generycznych. Między innymi zaproponowano obowiązek poinformowania krajowego urzędu rejestracji leków państwa, gdzie obowiązuje SPC oraz uprawnionego z SPC o zamiarze wytwarzania produktu w czasie trwania SPC nie później niż z 3 miesięcznym wyprzedzeniem oraz konieczność właściwego oznakowania produktu przeznaczonego na eksport5, co ma zapobiegać jego ewentualnemu przepakowaniu albo podrabianiu.

Powołując się na opracowaną na zlecenie Komisji Europejskiej analizę pod przewodnictwem Charles River Associates6 zmiany regulacji SPC mogą przynieść olbrzymie korzyści dla europejskiego rynku farmaceutycznego:
- 9,5 biliona euro dodatkowej sprzedaży netto,
- 25 tysięcy dodatkowych miejsc pracy,
- 3,1 biliona euro oszczędności dla europejskiego systemu ochrony zdrowia.
Niestety, na efekt zmian wynikających z przywileju" SPC MW producenci leków generycznych oraz pacjenci będą musieli poczekać.
Zakłada się bowiem, że SPC MW nie będzie obowiązywał dla leków aktualnie chronionych przez SPC, zaś dla preparatów, dla których złożono wnioski o przyznanie SPC przed zatwierdzeniem zmian w rozporządzeniu, a dla których SPC zostanie przyznane już po wprowadzeniu SPC MW, przyjęta cezura czasowa to 1 lipca 2022 lub 3 lata od daty wejścia w życie zmienionego rozporządzenia.
Projekt zmian do rozporządzenia nr 469/2009 czeka aktualnie na zatwierdzenie w Parlamencie Europejskim.
Footnotes
1. w tym artykule pojęcie generyki obejmuje także leki biopodobne.
2. http://www.spcwaiver.com/en
3. ibidem, infografika The Imapct of SPC Manufacturing Waiver on Jobs, Competitiveness &Patient Access to Medicines
4. https://data.consilium.europa.eu/doc/document/ST-6638-2019-INIT/en/pdf
5. Annex- I projektu zmiany rozporządzenia nr 469/2009, projekt logo umieszczanego na produktach przeznaczonych na eksport.
6. http://www.spcwaiver.com/en
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.
