
Last month, the UK MHRA published new guidance on human factors and usability engineering for medical devices to be taken into account when designing medical devices in accordance with the regulatory framework. 'Human factors' refer to how a person interacts with a product, and will depend on, among other things, the design of the product, the education and training of the intended user population, the environment in which they will be using the product, competing distractions, usability and ergonomics.
Human factors are increasingly being recognised as an important element of medical devices, in particularly in relation to managing patient safety. For example, a poor device design, with a complex user interface, may be associated with user-related design issues leading to problems with overdoses, or difficulties in drug administration, as illustrated by the following diagram from the guidance:
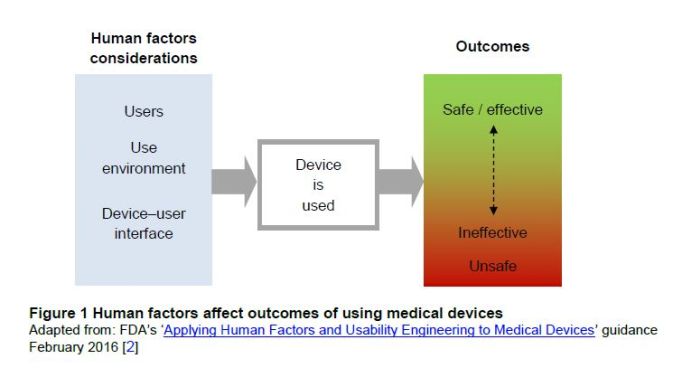
The aim of a usability engineering process and the inclusion of human factors into the design is to deliver products that are easy and safe to use. The guidance states that a well-designed product will be easy to use, and will have a user interface that is consistent with user experiences and expectations. Further, products designed with human factors in mind are more pleasing to use, and are therefore likely to lead to better adherence, and a greater chance of prescription.
Some of the human factors noted in the guidance, such as ergonomic design, are included in the essential requirements under the Medical Devices Directives, or the general safety and performance requirements under the new Medical Devices Regulations. Others signify best practice for regulatory expectations in the UK. The guidance sets out the typical stages of the design process, and highlights the iterative nature of design to take into account feedback from users during post-market surveillance. The approach taken should be risk-based, justified and appropriately documented in the technical documentation throughout the life-cycle of the product.
The guidance is intended for manufacturers of all device classes and of drug-device combination products, and notified bodies responsible for assuring the quality of those devices. It is intended to be advisory, rather than prescriptive, and to take into account international standards (such as IEC 62366), and guidance from the US Food and Drug Administration.
The content of this article is intended to provide a general guide to the subject matter. Specialist advice should be sought about your specific circumstances.